Animals and group assignment
In this study, 84 Wistar adult male rats, weighted 250-300 g, were selected randomly as subjects and assigned to 4 groups of sham, ischemia (given cell transplantation without ischemia), ischemia (given ischemia without cell transplantation), and transplant+ischemia (given cell transplantation and undergone ischemic surgery). In rats of the transplant recipient group, Sertoli cells were obtained from the other testicular tissue (from another rat). This group was subdivided into 4 subgroups in order to study infarct volume (n = 7), the permeability of the blood-brain barrier (n = 7), edema (n = 7), and the expression levels of NF-kB and Bax proteins (n = 7). In the ischemia group, the effect of cerebral ischemia, without cell transplantation, was assayed on infarct volume (n = 7), the permeability of the blood-brain barrier (n = 7), edema (n = 7), and expression levels of NF-kB and Bax proteins (n = 7). In the sham group, the effect of the stress of surgery and injection of Sertoli cell culture medium without ischemia and transplantation was evaluated (3 subgroups: permeability of the blood-brain barrier (n = 7), edema (n = 7), expression levels of NF-kB and Bax proteins (n = 7)). Due to the lack of ischemic induction, the infarction volume was not evaluated in the sham group. The effect of Sertoli cell transplantation without ischemia was considered in the control group (1 subgroup: expression levels of NF-kB and Bax proteins (n = 7)) (
Table 1).
The sham group is a non-ischemic group. This group was considered for the study of the effect of stress resulting from surgery and the effect of the infusion of Sertoli sulfate into the brain. Therefore, neurological defects and the other items should be investigated for comparison between sham, ischemia, and transplant+ischemia groups. But the control group was only considered as an evidence for the expression of inflammatory and apoptotic proteins.
In the transplant+ischemia group, the rats were subjected to 60 min of middle cerebral artery occlusion (MCAO) surgery 14 days after the injection of Sertoli cells. Twenty-four hours later, neurologic deficits, brain edema, infarct volume, and blood-brain barrier (BBB) integrity were evaluated.
Ethical Statement
This study was conducted in accordance with the rules of the National Institutes of Health and Care Guidance and the use of the Animal Laboratory (NIH Publications) revised in 2011, and the ethics committee of Shahid Beheshti University (No. 932,696). In this study, as little as possible, the animals have been used.
Death rates for groups: Control = 0, Sham = 13.3%, Ischemia = 33.3%, Transplant recipient = 20%.
Isolation and culture of Sertoli cells
Under approval by the animal care committee of Shahid Beheshti University of medical sciences, Wistar male rats (100-120 g) (n = 3) were killed under deep anesthesia (chloral hydrate, 800 mg/kg) and the testicles were removed. The tunica albuginea was removed from the testis and the tissue was sequentially digested first with trypsin (0.25%) (Gibco, USA) for 15 min and then with collagenase (0.1%) for 15 min at 37 °C. Afterward, fetal bovine serum (FBS) (Gibco, USA) was added and after centrifugation of the solution, the pellet was transferred to a culture media containing Dulbecco’s Modified Eagle’s Medium (DMEM/F12) (Gibco, USA), FBS (10%) and antibiotic. After 48 h, to remove the debris and red blood cells, the culture medium was changed (
9).
Immunocytoflouresent
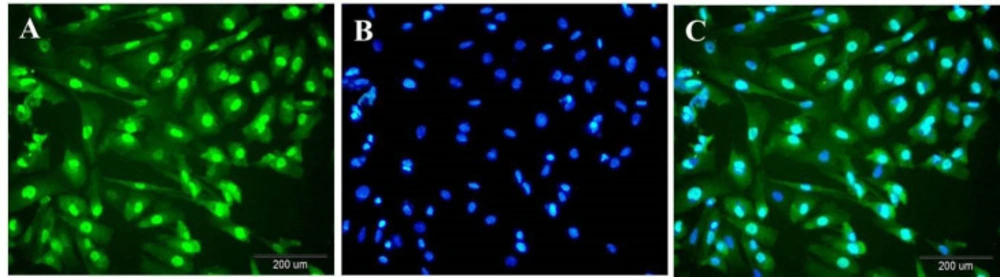
Sertoli cells were cultured in 24-well plate and fixed by 4% paraformaldehyde (PFA) (Merk, Germany). After washing with Phosphate Buffered Saline (PBS), cells were permeabilized with Triton Χ-100 (Sigma Aldrich, USA). Then, cells were incubated with goat normal serum followed by overnight incubation with the primary antibody against GATA4 (a member of the GATA transcription factor family) (Abcam, USA) at 4 °C. The fluorescent secondary antibody (goat anti-rabbit conjugated FITC (Abcam, USA)) was applied after washing with PBS. For visualization of the nuclei, the cells were stained with DAPI (Santa Cruz Biotechnology, USA). Preparations were examined under the fluorescent microscope (
14).
Sertoli cell transplantation
Unilateral Sertoli cell transplantation was performed in the right striatum. The striatum is associated with the cerebral cortex and the secreted trophic factors of Sertoli cells are absorbed by the striatum and sent to the cerebral cortex in the form of retrograde. The Sertoli cells were maintained alive in suspension using a 2 µL DMEM aliquot, stored on ice during the surgery procedure. After anesthetizing, the animals were unilaterally transplanted with 500.000 Sertoli cells in the right striatum labeled with DiI using a 5 µL Hamilton microsyringe placed at the following coordinates, relative to Bregma: +0.5 mm AP; −2.6 mm ML; −5 mm DV. Sertoli cells were transplanted in the part of the striatum (medio-posterior part) which was devoid of disruption of the extracellular matrix, in order to maximize graft survival. Based on the articles and also because the brain tissue does not have the capacity for more cells if more cells are injected, some injected cells are rejected (
15,
16).
Detection of the Sertoli cells after transplantation
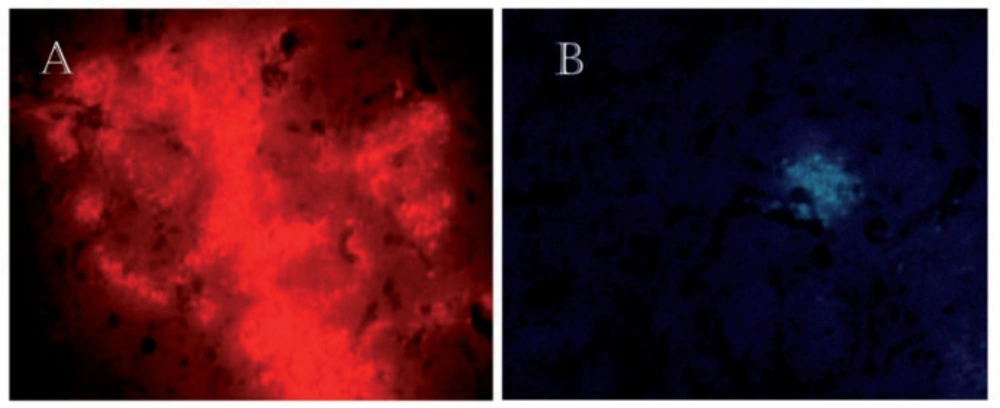
Before injection of the Sertoli cells, they were labeled with DiI (incubated with DiI, 5 μg/mL for 20 min before injection) and Hoechst (Sigma Aldrich, USA) (incubated with Hoechst, 4 μg/mL for one hour before injection) staining. After 14 Days, the rat was killed and the brain was prepared to determine the survival and distribution of the transplanted cells. Detection of these cells were carried out by fluorescing microscopy.
Induction of cerebral ischemia by the MCAO method
Fourteen days after the recovery (Time to show trophic effects of Sertoli cells), the rats were anesthetized to undergo an intercostal artery occlusion surgery. For this purpose, a nylon suture (3-0) was inserted into the internal carotid artery through the trunk of the common carotid artery to reach the anterior carotid artery. As a result of the insertion of the suture and blockage of the arterial blood flow pathways, the blood flow from each side was obstructed to the middle cerebral artery (MCA). After 60 min of ischemia, the blood flow was restored. The body temperature was measured through a rectum digital thermometer and maintained at 37 °C using a thermal pad (
17).
Neurologic deficits Score
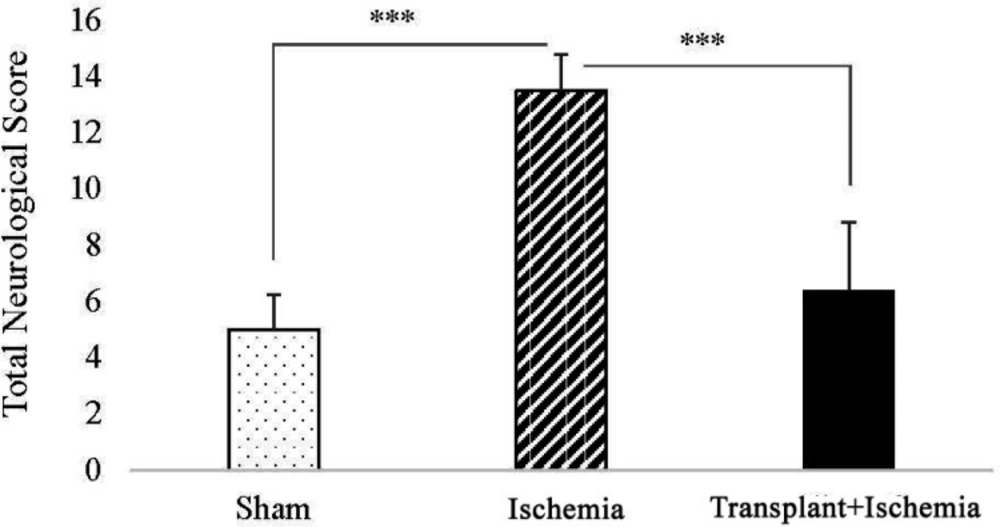
Twenty-four hours after the induction of ischemia, behavioral tests were performed on the rats to evaluate neurologic deficits. Neurological tests were conducted in five categories of Raise the Tail, Sensory Function, Motor Function, Beam Test and reflex activity (the minimum and maximum scores on these tests were 0 and 18, respectively) (
18).
Infarct volume
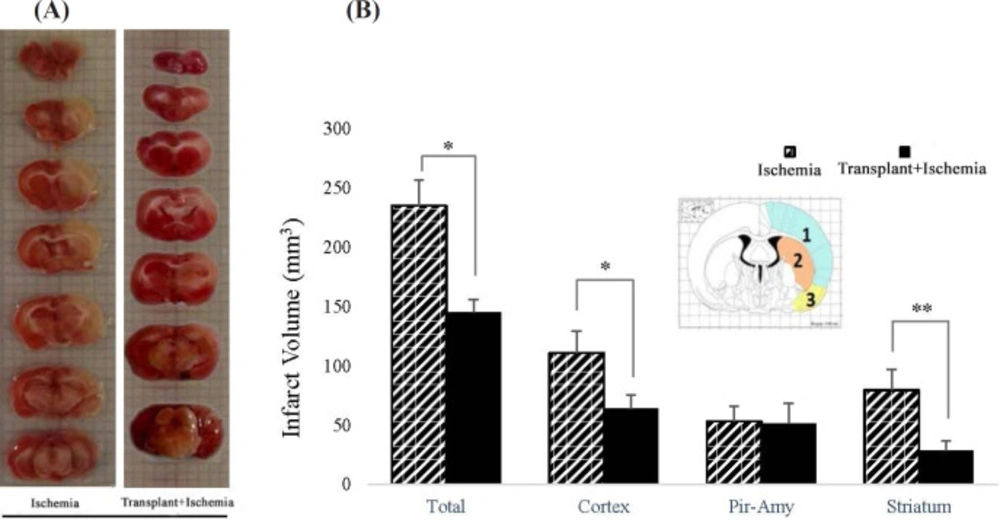
After performing neurological tests and killing the rats, their brain was removed and kept in cold saline at 4 °C for 5 min. Then, coronal brain slides with a thickness of 2 mm were prepared using the brain matrix. The brain slices were put in 2% TTC, 2,3,5-triphenyl tetrazolium chloride solution, solution (Merck, Germany) at 37 °C for 15 min to be stained. Then, some photos were taken from these slides using a digital camera (Canon EOS 500D digital). Finally, the area of the ischemic site in each slide was measured using Image J (Version 1.50) and calculated using the Swanson method based on the following equation. In this part, infarct volume was analyzed in the whole hemisphere, cortex, piriform cortex-amygdala, and striatum separately. The brain areas were identified using the Rat Brain Atlas of Paxinos and Watson (
19).
The corrected volume of damaged area = Left Hemisphere Volume – (Right Hemisphere Volume – Damaged Area Volume).
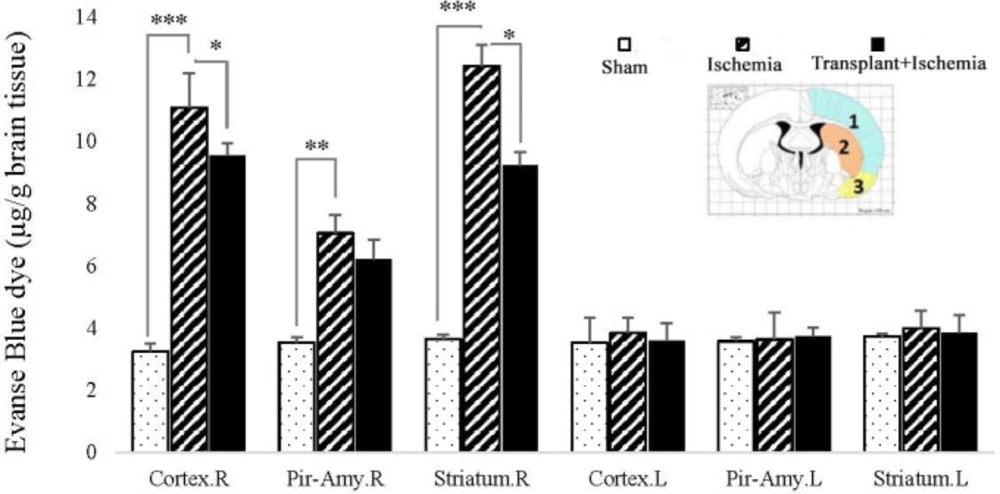
The permeability of the blood-brain barrier
The integrity of the blood-brain barrier was evaluated by measuring the amount of Evans Blue dye (EBD) (Sigma Chemicals, USA) extravasation. First, 30 min after the onset of ischemia, the rats were treated with 4 mL of 2% Evans Blue per body weight intravenously. Twenty-four hours after the induction of ischemia, the rats were anesthetized and intravascular Evans blue was replaced with saline using the transcardial method. Then the brain was removed and its different parts (cortex, piriform cortex-amygdala, and striatum) were separated in each hemisphere and weighted. To measure the EBD extravasation, the brain tissue was homogenized in phosphate buffer and 60% trichloroacetic acid was added to the solution to precipitate the protein. Then, the microtubes were vortexed for 2 min and kept in a fridge at 4 °C for 30 min. In the next stage, the samples were centrifuged at 1000 rpm for 30 min. Finally, the EBD absorbance in the supernatant was measured at 610 nm wavelength by a spectrophotometer (Perkin-Elmer, Illinois, USA) and its concentration was calculated according to the standard curve and expressed as μg/g of brain tissue (
20,
21).
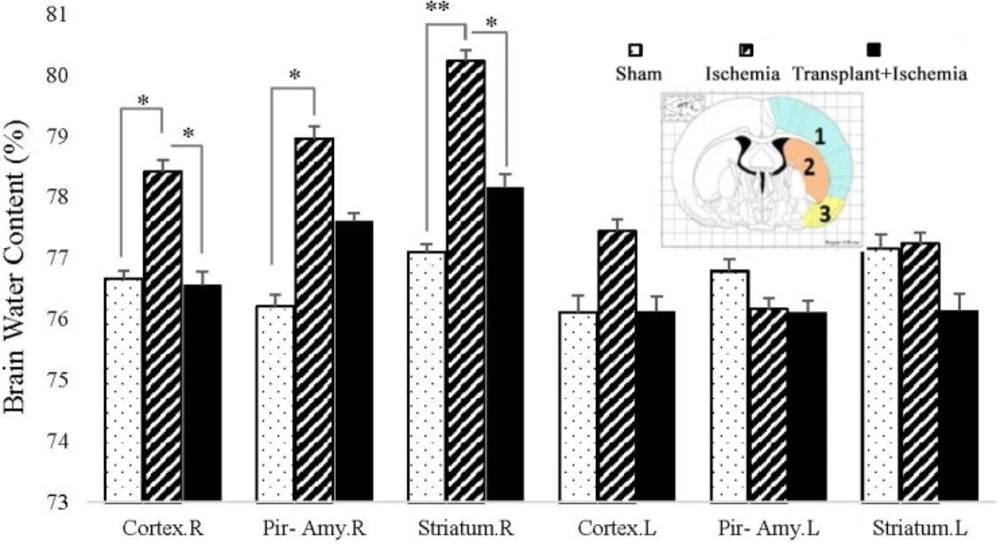
The water content of the tissue
After decapitation of rats, their brain was removed and the cerebellum, pons, and olfactory bulbs were separated. Then, the wet weight (WW) of different parts of the brain (cortex, piriform cortex-amygdala, and striatum) was assessed. After drying them in an oven at 120 °C for 24 h, their dry weight (DW) was also measured. Finally, the water content of the brain was calculated using the following Equation (
20): [(WW−DW)/WW] × 100
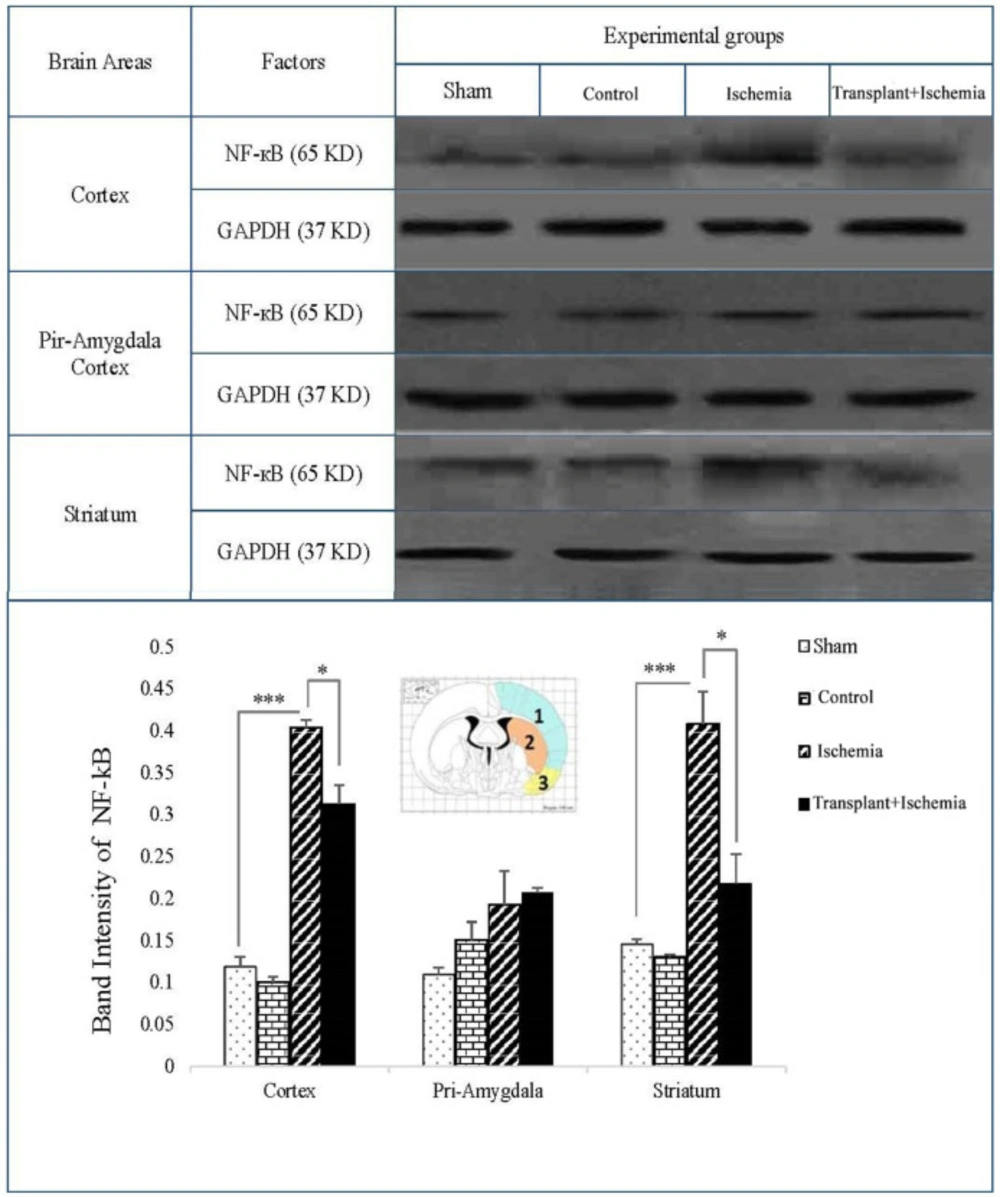
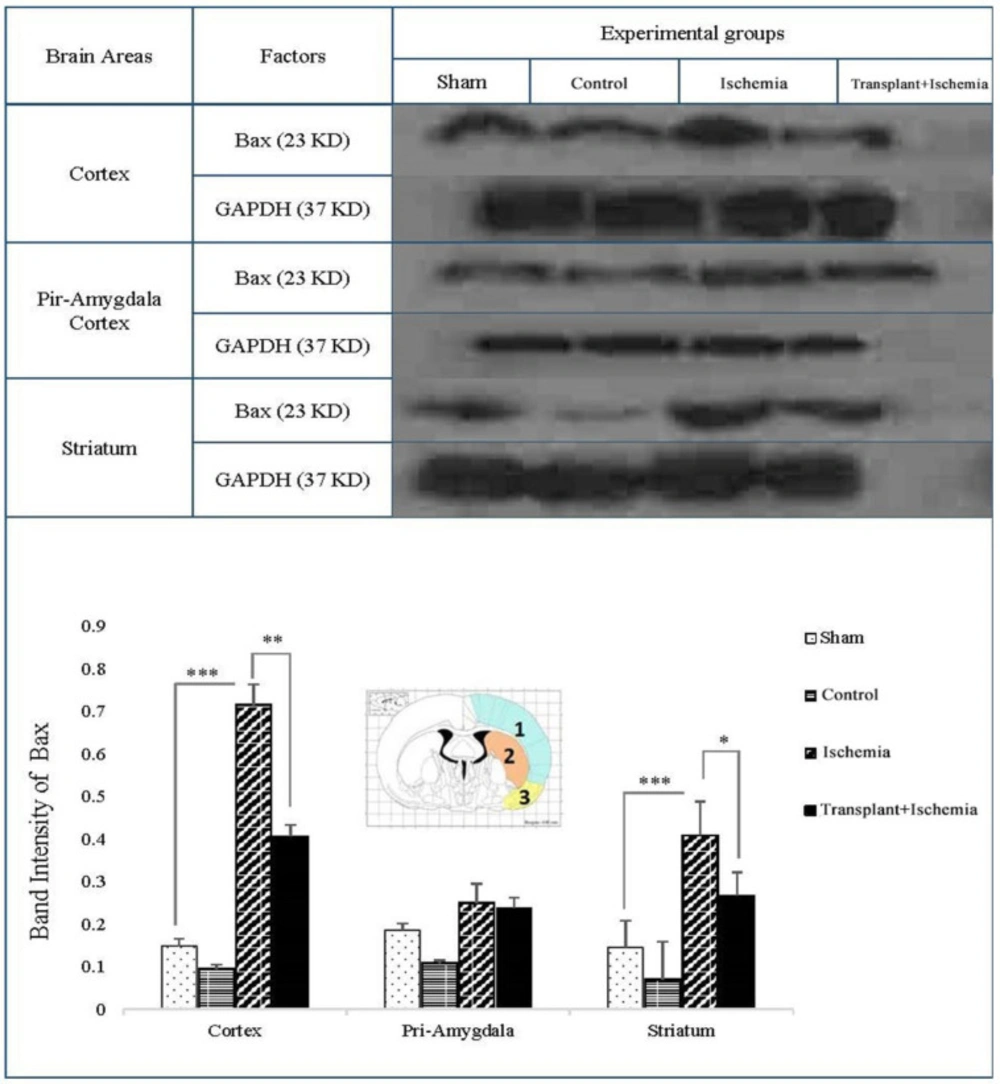
Western blot
The studied brain zones (cortex, piriform cortex-amygdala, and striatum) for each group (sham, control, ischemia, and transplant+ischemia) were identified and isolated using the Rat Brain Atlas of Paxinos and Watson. Equal volumes of obtaining samples to prepare the protein extract were homogenized and centrifuged with a lysis solution (Tris-Hcl_SDS_EDTA_NaCl_Sodium Deoxy cholate_Protease inhibitor cocktail_NP40 (0.1%)) and the supernatant was achieved. Supernatants were transferred to the gel using the electrophoresis gel. Proteins were separated by SDS-PAGE (10% gel) and then transferred to the PVDF membrane (Millipore) using a Western tank containing a transfer buffer (Tris_Glycine_Methanol 20%_Distilled Water). Blocking of blots was performed by using a blocking solution containing 2% non-fat dry milk in Tris buffer saline in 0.1% Tween 20 at 4 ºC for 75 min. Then, the blots were incubated with rabbit anti-NF-кB polyclonal (1:500 dilution; Santa Cruz), rabbit anti-Bax polyclonal Antibody (1:500 dilution; Santa Cruz, USA) and rabbit anti-β-actin (1:1000 dilution; Santa Cruz) antibodies, followed by goat anti-rabbit secondary antibody (1:500 dilution; Santa Cruz) for 90 min separately. Finally, the desired proteins were identified using advanced chemiluminescence (Enhanced Chemiluminescence, Amersham Biosciences) and film exposure. The signal density of the blots was measured by an image analysis system (ImageJ, version 1.46r).
Statistical analysis
Data analysis was performed in SPSS (v22.0). Data from neurologic deficits were analyzed using Kruskal-Wallis followed by the Dunn’s test and the statistical analysis of data obtained from infarct volume, was done by non-parametric Mann-Whitney U test (SPSS v22.0). Moreover, brain edema, blood-brain barrier permeability, and expression of NF-kB and Bax proteins were compared using two-way analysis of variance (ANOVA) (SPSS v22.0 post-hoc LSD). The results were reported as mean ± SEM and the level of significance was determined to be (P < 0.05).