General experimental procedures
High-performance liquid chromatography (HPLC) was done on a Waters HPLC 501 equipped with ultraviolet (UV) and refractive index (RI) detectors and YMC Pack-Sil normal HPLC column (250 × 20 mm, YMC, Japan). The nuclear magnetic resonance (NMR) spectra were captured using an Avance AV400 (Bruker, Germany). Thin layer chromatography was done on Merck silica gel alufoils and visualized by natural flavonoid product (NP) reagent (1% of 2-aminoethyl diphenylborinate in methanol) and ceric sulfate (1 g ceric sulfate, 50 mL H2SO4, and 500 mL water). Column chromatography was done using Silica gel adsorbents (63-200 μm; 40-63 μm, Merck, Germany) and Polyamide SC6 (Machery Nagel, Germany). Chloroform (ChCl3) and methanol (MeOH) HPLC grade solvents were purchased from Caledon (Canada), and other solvents from Pars chemistry company, Iran, otherwise it is determined in the text.
Cancer cells were obtained from National Cell Bank of Iran (NCBI). Well plate absorbance (OD) was read using ELISA microplate reader (Bio-Rad, Hercules, CA, USA). The Annexin V-FITC/PI stained cancer cells were counted by a FACS Calibur flow cytometer (BD Bioscience, USA). The amplification of interested gene was performed by Applied Biosystems instrument (ABI 7500 Real-Time PCR System, Foster City, USA). The fluorescence of ThT-protein aggregates was measured at 485/535 nm using Synergy H1 Multimode Microplate Reader.
Plant material
Aerial parts of C. schmidii Wagenitz (Asteraceae) was collected from North Khorasan state of Iran at summer at elevation of 1920 m. Plant material was identified by Mohammad Reza Joharchi, Herbarium of Ferdowsi University of Mashhad, Mashhad, Iran where a voucher specimen (FUMH. 45068) was deposited.
Extraction and isolation
Plant material was dried in shade condition (2200 g), macerated in acetone (10 L) at ordinary temperature for five days, and repeated three times. The resulted extract was concentrated using a rotary evaporator (Heidolph Instruments, Germany) at 40 ºC (230 g). The concentrated extract was partitioned between hexane and aqueous methanol (70:30) in a separating funnel to remove fats and chlorophylls. Methanol partition was concentrated and subjected on silica gel gravity column using a stepwise gradient of hexane: acetone (Fr.1, 90:10; Fr.2, 80:20; Fr.3, 70:30; Fr.4, 50:50; Fr.5, 0:100). Fr.5 with flavonoid pattern in the TLC profile was subjected on polyamide SC6 column using ChCl3: MeOH (Fr.5a, 90:10; Fr.5b, 85:15; Fr.5c 83:17; Fr.5d 80:20). FR.5b, and Fr.5c with the TLC profile of flavonoids were purified by HPLC pump on a silica gel column using ChCl3: MeOH (80:20), with a flow rate of 3 ml/min using RI detector and UV detection at 250 nm (
Figure 1).
Spectral data
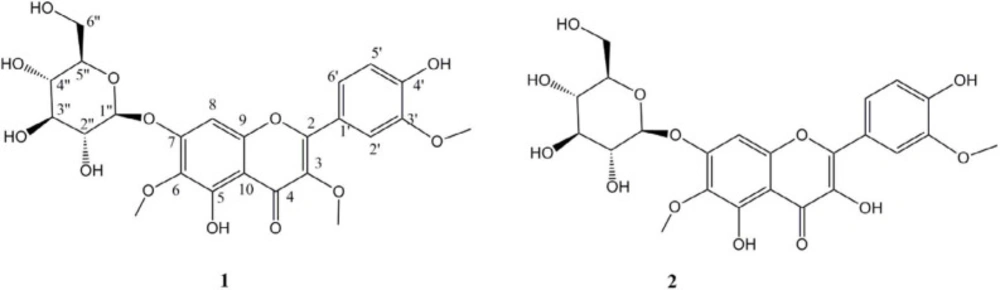
Compound 1 (21 mg): 1H-NMR (400 MHz, Dioxane D) δ: 3.2-3.8 (H-2״- H-6״), 3.73 (3H, s, OMe-6), 3.77 (3H, s, OMe-3), 3.84 (3H, s, OMe-3׳), 4.97 (1H, d, J = 7.6, H-1״), 6.67 (1H, s, H-8), 6.87 (1H, d, J = 8.4, H-5׳), 7.54 (1H, dd, J = 2, 8.8, H-6׳), 7.63 (1H, d, J = 2, H-2׳). 13C-NMR (100MHz, Dioxane D) δ: 55.4 (OMe-3׳), 59.3 (OMe-3), 59.9 (OMe-6), 61.3(C-6’’), 69.6(C-4’’), 73.5(C-2’’), 77.1(C-3’’), 77.3(C-5’’), 93.7(C-8), 100.7(C-1’’), 107.2(C-10), 111.5(C-5’), 115.2(C-2’), 122.0(C-1’), 122.4(C-6’), 133.1(C-6), 138.4 (C-3), 147.0(C-3’), 149.2(C-4’), 151.7(C-9), 153.3(C-5), 155.8(C-2), 156.2(C-7), 178.8 (C-4). ESIMS molecular ion of [M-H]- at 521 m/z.
Compound 2 (15 mg): 1H-NMR (400 MHz, Dioxane D) δ: 3.4-3.7 (H-2״- H-6״), 3.85 (3H, s, OMe-3׳), 3.94 (3H, s, OMe-6), 5.11 (1H, d, J = 7.6, H-1״), 6.83 (1H, s, H-8), 6.98 (1H, d, J = 8.4, H-5׳), 7.76 (1H, dd, J = 2, J = 8.4, H-6׳),7.87 (1H, d, J = 1.6, H-2׳). 13C-NMR (100MHz, Dioxane D) δ: 55.3 (OMe-3׳), 60.0 (OMe-6), 61.3(C-6’’), 69.6(C-4’’), 73.5(C-2’’), 77.1(C-3’’), 77.3(C-5’’), 93.9(C-8), 100.8(C-1’’), 105.5(C-10), 110.9(C-5’), 115.3(C-2’), 121.8(C-6’), 122.7(C-6’), 132.5(C-6), 136.2 (C-3), 146.4(C-3’), 147.1(C-4’), 145.5(C-9), 151.6(C-5), 152.2(C-2), 156.4(C-7), 146.1 (C-4). ESIMS molecular ion of [M-H]- at 507 m/z.
Cell culture
Two different human breast cancer cell lines, MCF-7 as estrogen receptor positive, and MDA-MB 231 as estrogen receptor negative, were obtained from National Cell Bank of Iran (NCBI). The cells were cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS), contained penicillin (100 u/mL), and streptomycin (100 μg/mL), in a 5% CO2 incubator at 37 °C (
9).
Cell viability assay
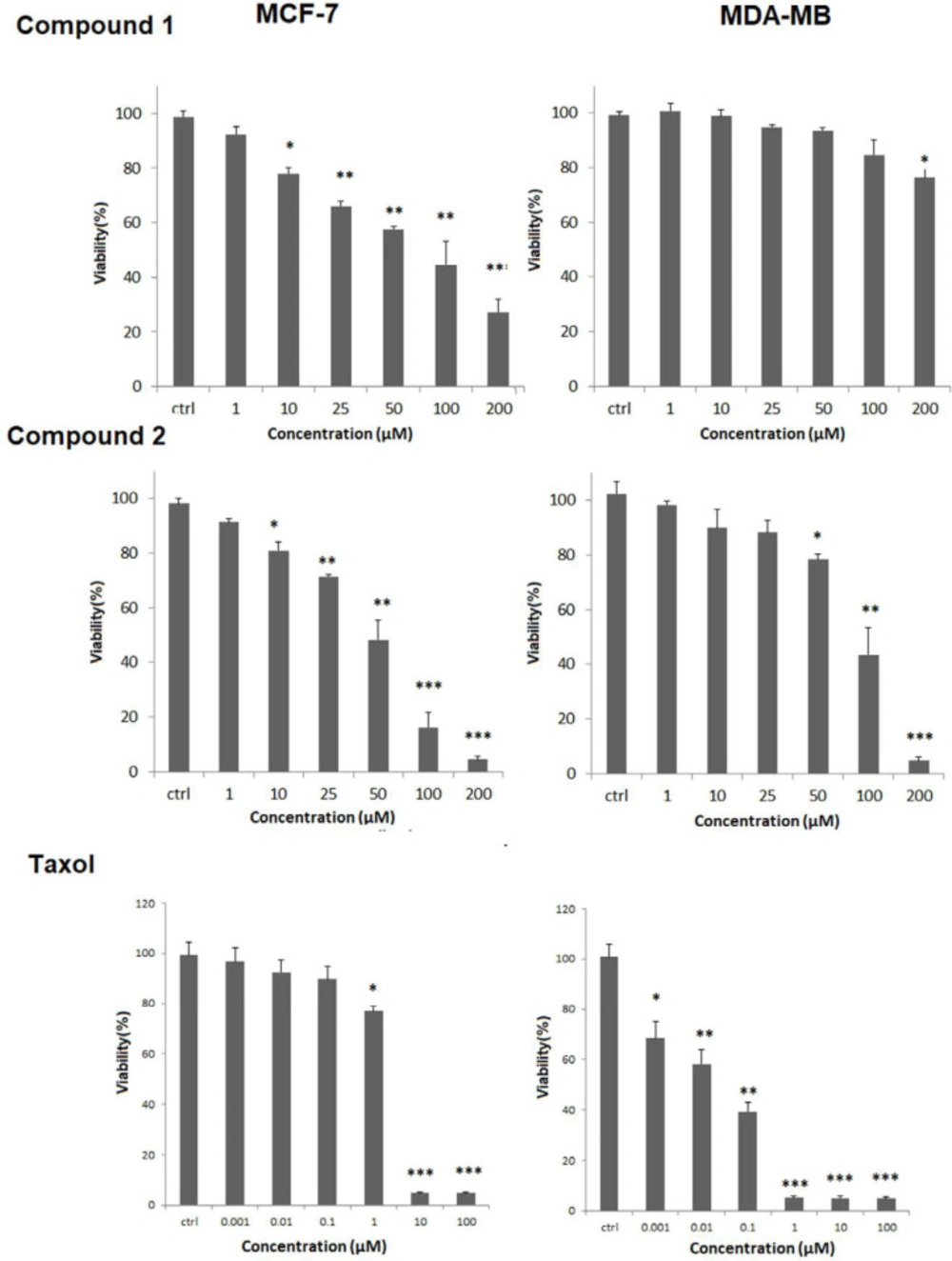
Cell viability was determined by MTT assay as described previously (9). Briefly, the cells with a density of 5 ×10
3 cells /well were seeded into 96-well plates in RPMI media and incubated overnight (5% CO2, 37 °C). Then new media was replaced, and tested compounds
1-2 in the concentrations of 1, 10, 25, 50, 100, 200 μM, Taxol (Ebewe Pharma, Austria) in the concentrations of 0.001, 0.01, 0.1, 1, and 10 μM, and corresponded solvent for each concentration as negative control were added and incubated for next 48 h. Then 20 μL of MTT (5 mg/ml in PBS) was added to wells incubated again for 4 hr. at the same conditions. Then supernatants were eliminated, biological grade DMSO was added, and the absorbance (OD) was read at 570 nm in an ELISA microplate reader (
9).
Annexin V-FITC/PI assay of apoptosis
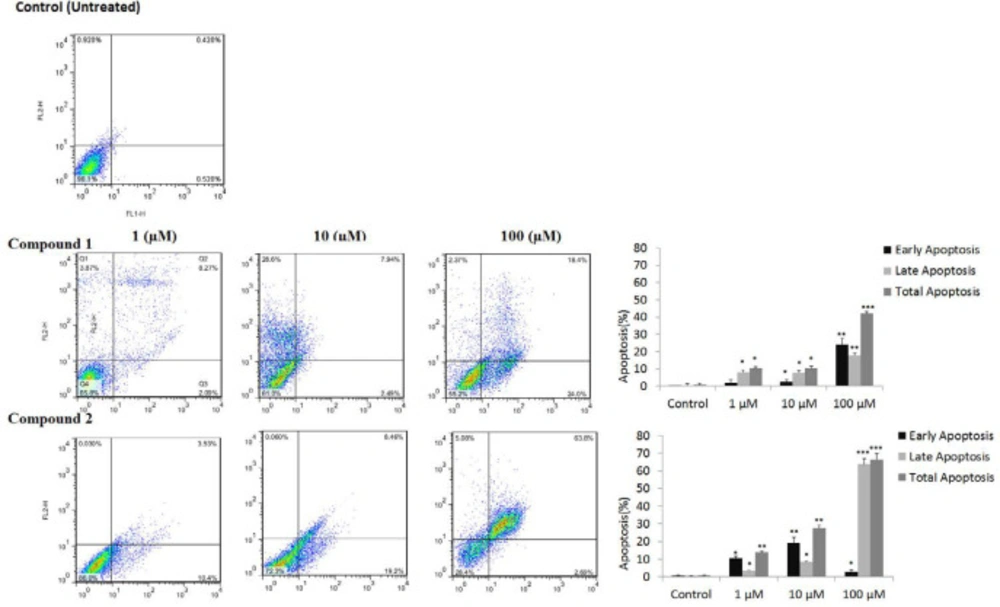
Apoptotic cell death induced by compounds was quantified by flow cytometry using the Annexin V-FITC/PI kit (
10). Briefly, MCF-7 cells were seeded in a six-well plate by the density of 3×10
5 per well and treated with tested compounds (0.1, 1, 10, and 100 µM) for 24 h in an incubator (5% CO2, 37 °C). Floated and attached cells were washed twice with PBS and suspended again in binding buffer. Then using Annexin V-FITC/PI kit protocol, Annexin V-fluorescein isothiocyanate/ PI (each 5 µL) were added to wells to stain the cells and incubated again for 10 min at room temperature. The stained cancer cells were counted by a FACS Calibur flow cytometer, and the results analyzed using the software in the instrument (
10).
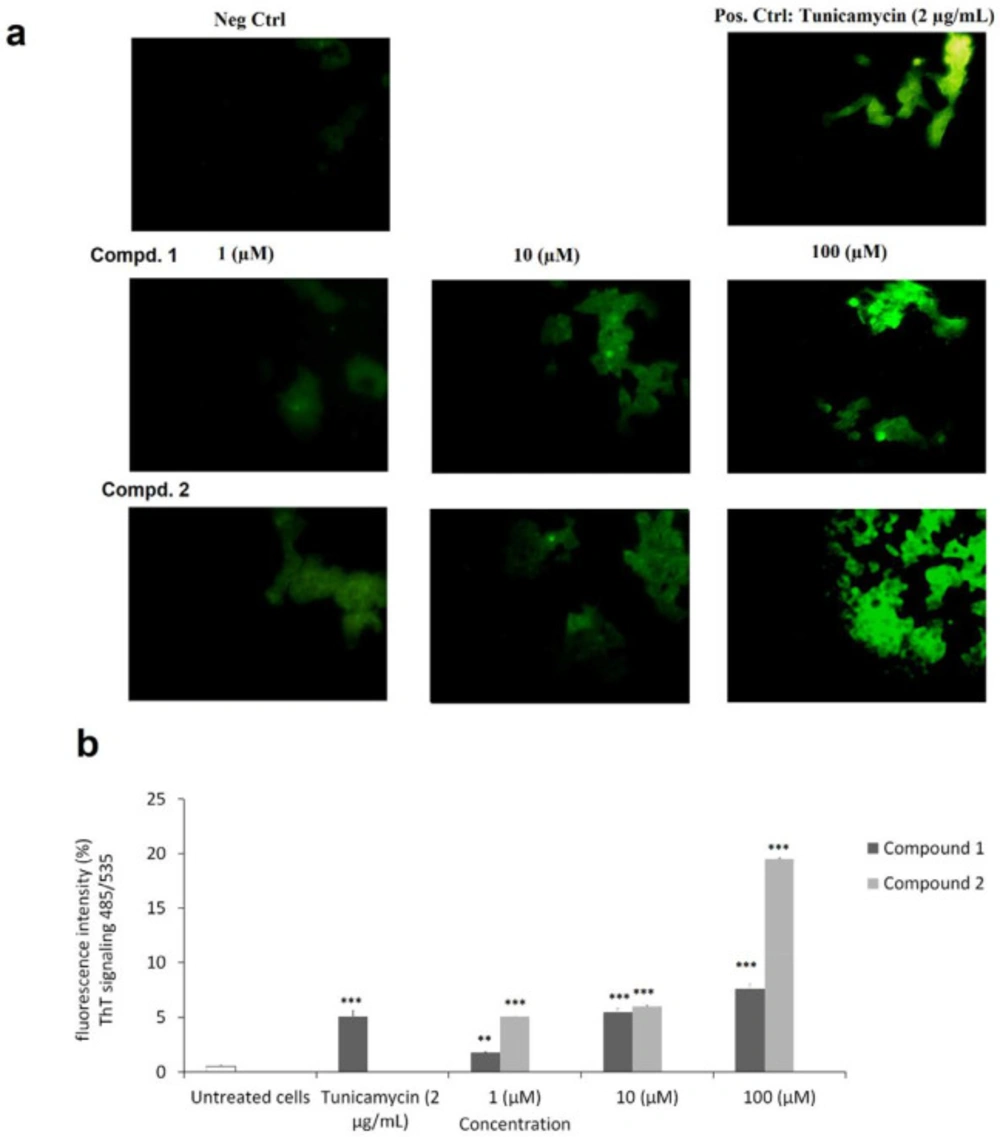
Endoplasmic reticulum stress assay by Thioflavin staining and fluorescence microscopy analysis
ER stress detection by Thioflavin T (ThT; Sigma Aldrich, USA) staining of unfolded and aggregated proteins was used to check MCF-7 cellular ER stress. MCF-7 cells were seeded in a density of 3 × 10
5 per well in a 24-well plate and incubated by adding tested compounds in the concentrations of 1, 10, and 100 µM, and Tunicamycin (2 μg/mL) as positive control for 24 h. Untreated cells were regards as negative control. ThT (5mM) was added to wells, and after half of an hour incubation (37 °C), Triton in PBS (0.1%) was added, and using a cell scraper, cells were lysed. Cell lysates (100 mL) in each well were transferred into a black-bottomed well plate special for the ELISA fluorescence reader, and examined ThT stained protein aggregates at 485 nm excitation and 535 nm emission spectral condition. ImageJ software was used to analyze, and measure the relative fluorescent signals (
10).
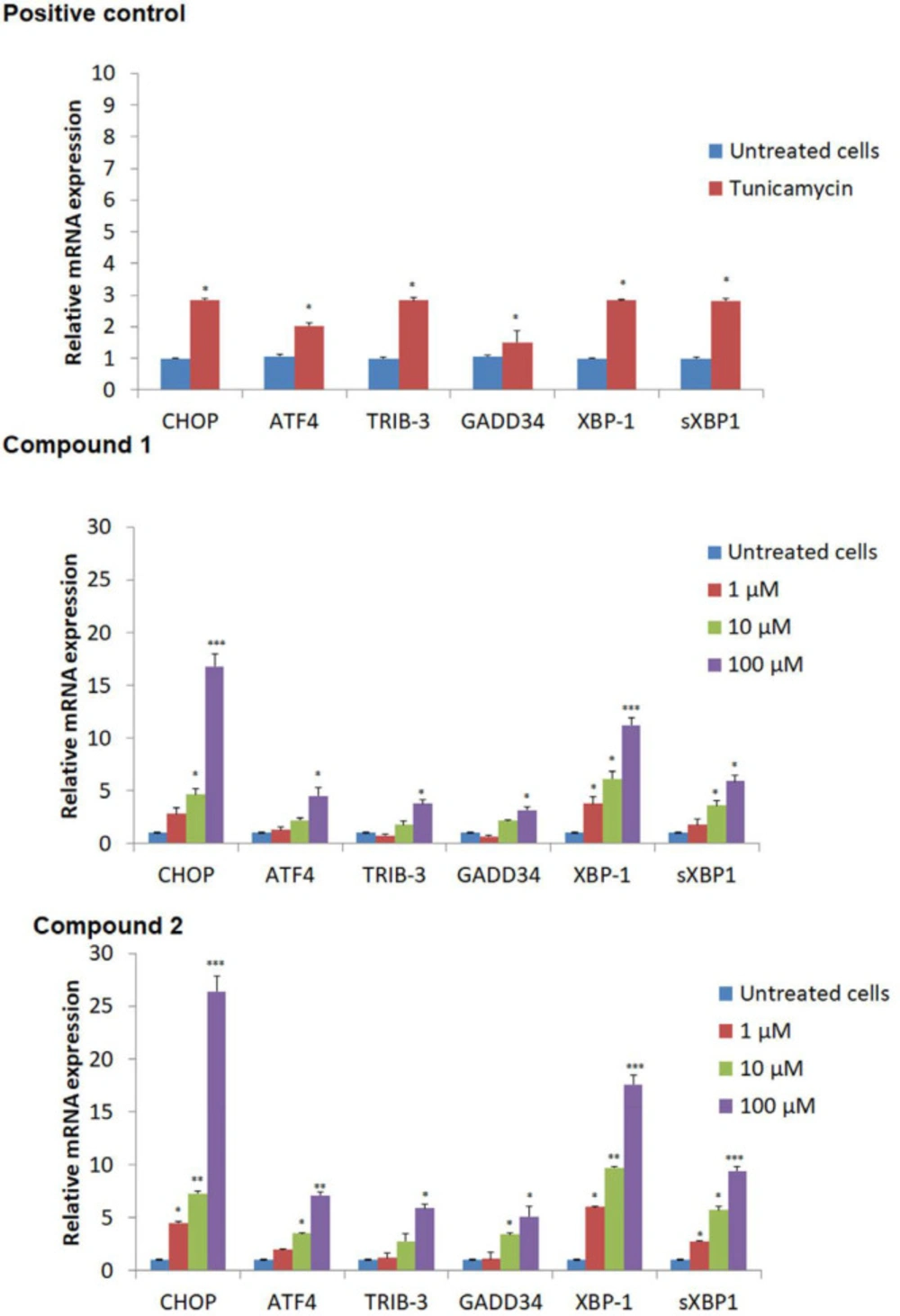
ER Stress Gene Expression
ER stress gene expression was done as described before (10). Briefly, MCF-7 Cells were exposed for 24 h to tested compounds
1-2 in the concentrations of 1, 10, and 100 μM, Tunicamycin (2 μg/mL) as positive, and using GAPDH (Glyceraldehyde 3- Phosphate Dehydrogenase) as an internal control. Total RNA was extracted by Trizol (Sigma-Aldrich, USA), and its purity and concentration were checked by UV spectrometer. RNA reverse transcription was done using Qiagen kit (Qiagen, UK), and cDNA was extracted and subjected to real-time quantitative PCR (50 °C for 2 min, 95 °C for 10 min, 40 cycles at 95 °C for 15 s and 60 °C for 1 min). The sequences of XBP-1, sXBP-1, ATF-4, TRIB-3, GADD34, and CHOP primers are mentioned in
Table 1. Using Excel software, all the real-time gene expression levels were normalized and validated by GAPDH. Finally, the mRNA expressions were reported using the 2
-ΔΔCt analysis (
3).
In-silicoanalysis
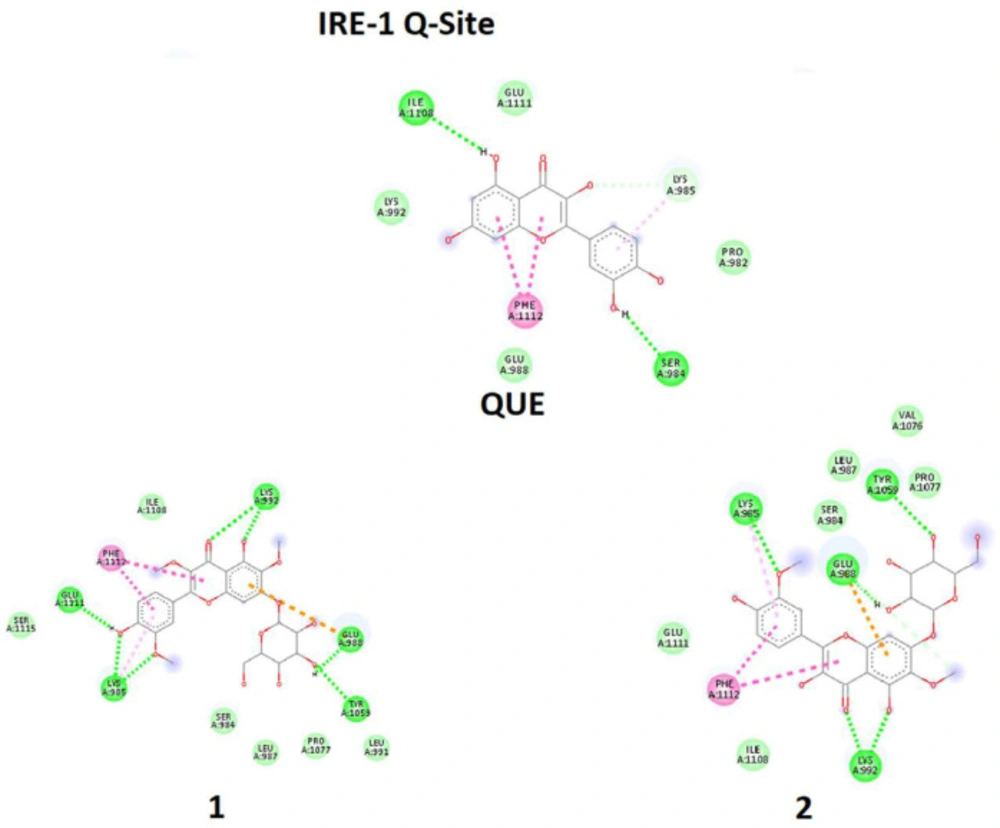
IRE1α is a protein with RNase function which cut X-box-binding protein 1 (XBP1) mRNA in response to ER stress and made its active spliced form (sXBP1), leading to UPR (
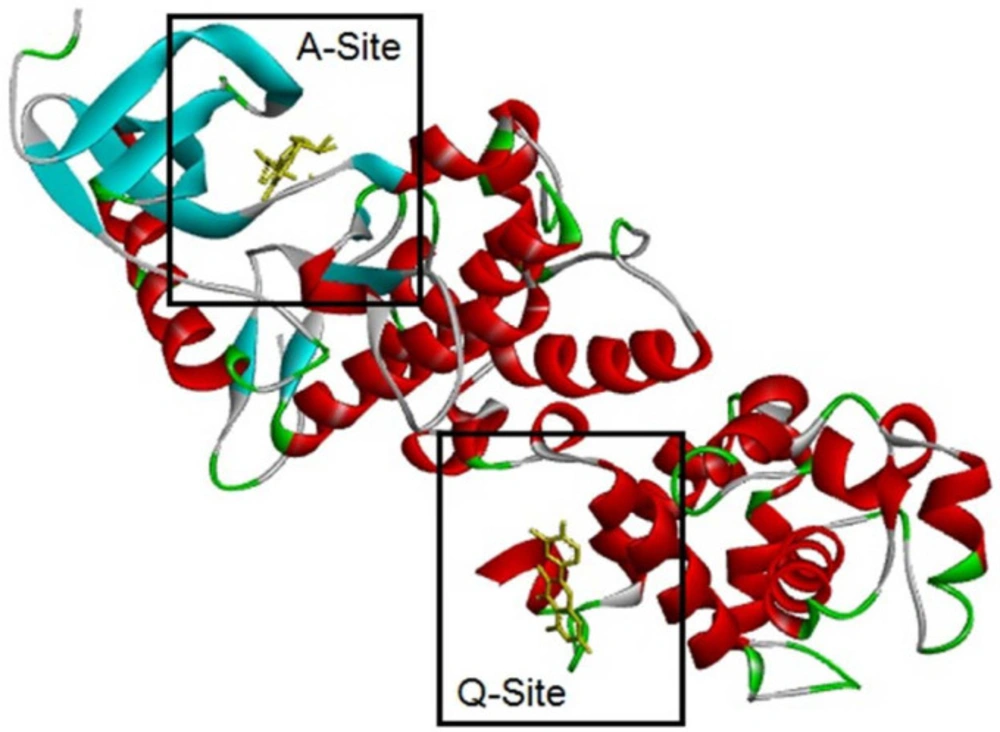
2). Fohlen
et al. showed that XBP1 splicing is done only by IRE1α. Therefore, the marked increase in sXBP1 in this study encouraged us to check the binding affinity of these compounds on active sites of IRE1. IRE1 has one active site named A-site and another binding site for substrate mRNA (
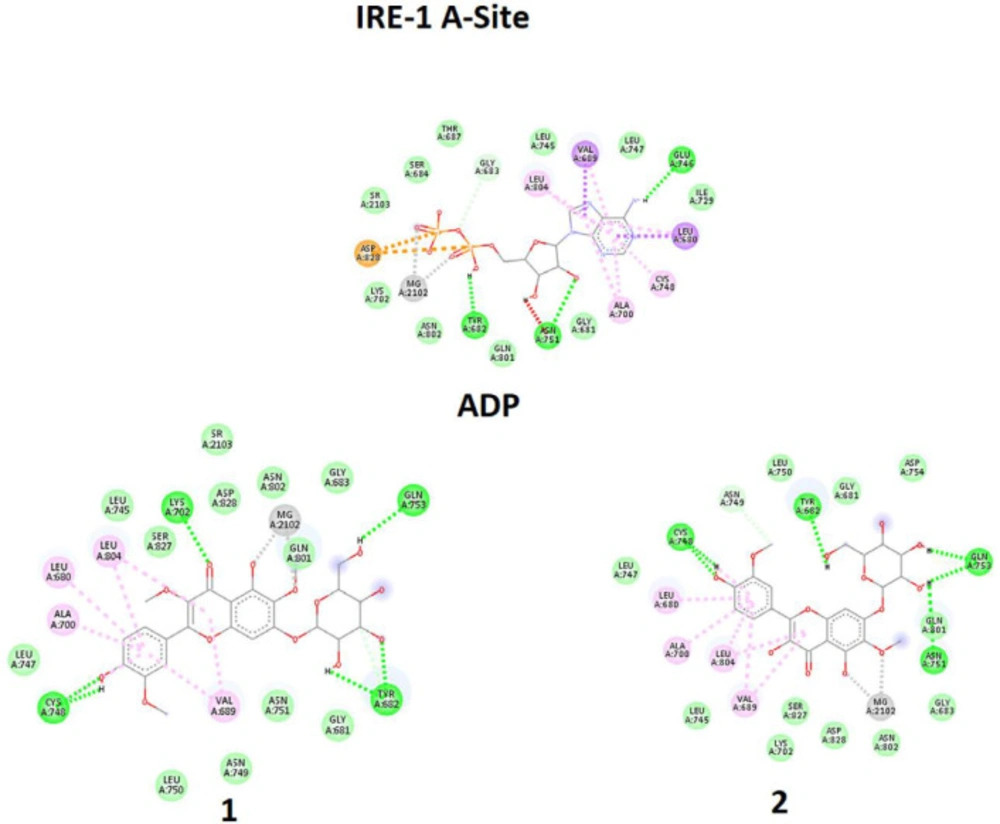
11) required for RNase activity. Adenosine diphosphate (ADP) can bind to A-site as an indigenous endoribonuclease activator of IRE1 (
12). Luke Wiseman et ql. reported that quercetin (QUE) with flavonol structure bind on another binding site named Q-site in the presence of ADP in A-site. They made a co-crystal of IRE1 with ADP in A-site and quercetin in Q-site and reported that quercetin enhances ADP activity while stabilizing also a dimeric form of IRE1 (
13). Compound
1-2 also has a flavonoid structure, and considering it, we performed computational docking and
in-silico analysis to check if tested flavonoids can act like QUE on IRE1 and enhance RNase activity. In the first, the model structures have been optimized to the minimum energy by the Gaussian 09 program (
14) under the B3LYP/3-21G* standard theoretical level of density functional theory (DFT). To approve non-imaginary frequency existence, frequency calculations have been done at the same theoretical level, and the structures have been found valid. As a result, three ligands have been prepared for further investigations by the Molecular Docking simulations (MDs). In the next step, the 3LJ0 PDB structure of IRE1 complexed with adenosine diphosphate (ADP) and Quercetin (QUE) (
13) was obtained from Protein Data Bank (id: 3LJ0), and it was prepared as a target for MDs by eliminating ADP, and QUE. Furthermore, the Kollman charge has been added to the hydrogenated PDB. The grid box has been set to 40 × 40 × 40 once for docking of A-Site and once again for docking of Q-Site with assigned 100 numbers of genetic algorithm (GA) conformational search as implemented in the AutoDock4 program for each docking process. In addition, Gasteiger charges were added to each of the ligand structures to be prepared for MDs processes (
14).