Materials and Methods
Site-directed mutagenesis and Construction of the recombinant expression plasmids
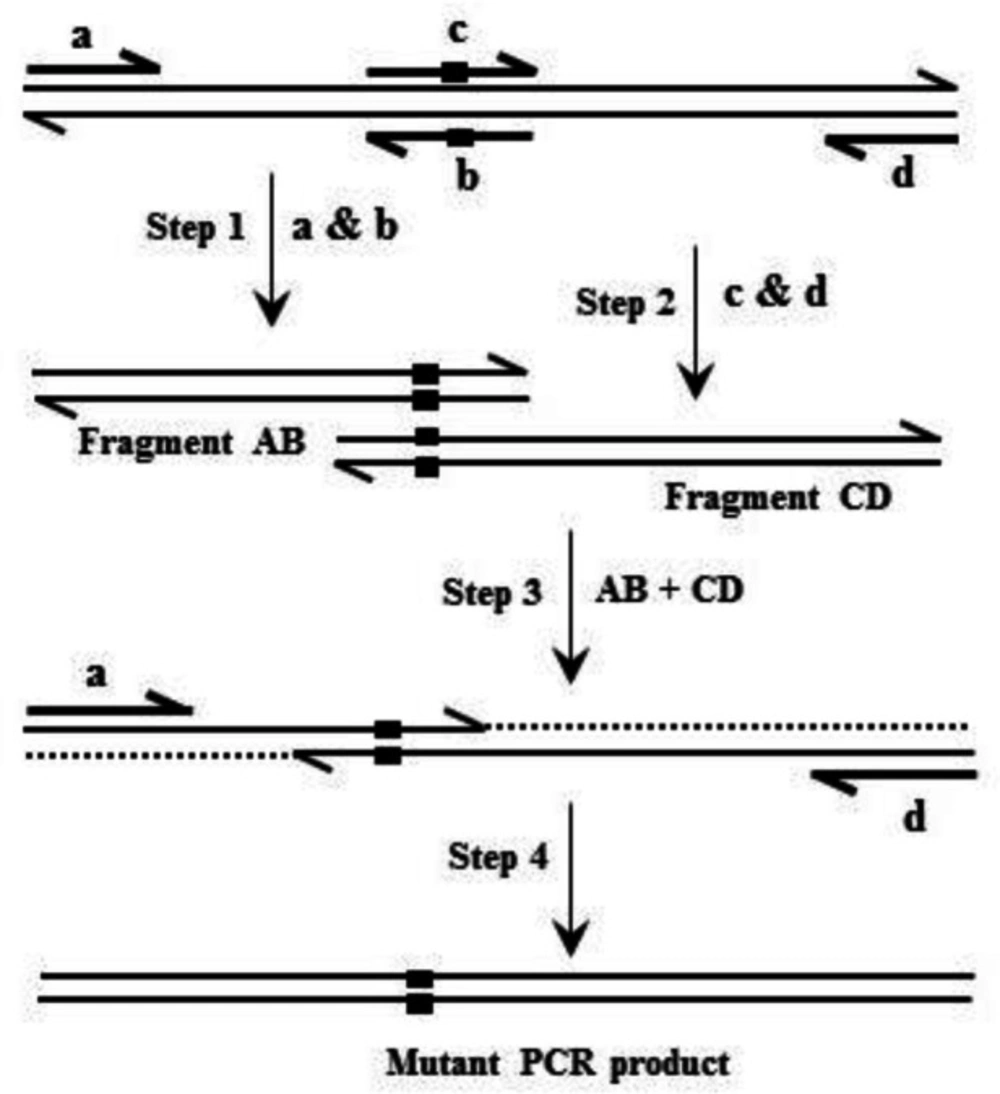
Based on overlap extension PCR (
Figure 1), site-directed mutagenesis was applied to introduce mutations into the hFIX cDNA (
28,
29). Oligonucleotides used for site-directed mutagenesis are listed in
Table 1. Oligonucleotide pairs K22N-F/K22N-R and R37N-F/R37N-R were used respectively for introducing K22N and R37N substitutions into hFIX cDNA. Oligonucleotides hFIX-
EcoRI and hFIX-
BamHI equipped respectively with
EcoRI and
BamHI restriction sites, were used as forward and reverse primers for amplification of the pre-pro hFIX or its N-glycosylation mutants’ coding sequences. Primer hFIX-
BamHI was equipped with a nucleotide sequence of 6xHis before the stop codon in order to introduce a His-tag to the recombinant proteins’ C-termini. The amplified fragments were then cloned into the pIRES2-EGFP vector, between
EcoRI and
BamHI restriction sites, upstream to the IRES-EGFP sequence.
Following NheI/NotI digestion, the target fragments, comprising the coding sequences of either the hFIX or its mutants and IRES-EGFP sequence, were subcloned into the pCEP4 expression vector (Thermo Fisher Scientific, USA), downstream to the cytomegalovirus (CMV) immediate early promoter/enhancer to end up with three bicistronic recombinant plasmids namely; pCEP4-hFIXwt, pCEP4-hFIXK22N, and pCEP4-hFIXR37N.
Cell Culture, Transfection, and Transient Expression
Suspension adapted Chinese hamster ovary (CHO-s) cell line (Thermo Fisher Scientific, USA), considered as a mammalian expression host, was cultured in FreeStyle™ CHO Expression Medium (Thermo Fisher Scientific, USA) supplemented with 8mM L-glutamine (Thermo Fisher Scientific, USA) and incubated at 37oC in an 8% CO2 atmosphere while shaking at 140 rpm. For transient expression, the cells were transfected with bicistronic recombinant plasmids; pCEP4-hFIXwt, pCEP4K22N, and pCEP4R37N (expressing either wild-type or mutant forms of the hFIX, together with an enhanced green fluorescent protein, EGFP, as a reporter protein), using FreeStyle™ MAX reagent (Thermo Fisher Scientific, USA), according to the manufacturer’s instruction. Three to five h after transfection, vitamin K (Caspian Tamin, Iran) was added at a final concentration of 2.5µg/mL. At 24, 48, and 72 h post-transfection, the cells were pelleted and the culture media were harvested and subjected for expression analyses.
Transcript Stability analysis
Secondary structures of the mRNAs from complete sequences of the hFIX
wt and its N-glycosylation mutants’, were predicted using Mfold Web Server (http://unafold.rna.albany.edu/?q=mfold) (
30). The analyzed regions included the coding sequences of the hFIX
wt or its mutants and the IRES-EGFP sequence.
RNA Extraction and cDNA Synthesis
Total RNAs were isolated from the transfected CHO-s cells at 24, 48, and 72 h post-transfection using high pure RNA isolation Kit (Roche, Germany), according to the instruction provided by the manufacturer. Both quantity and purity of the total isolated RNAs were determined by UV absorbance measurement at 230, 260, and 280 nm by NanoDrop spectrophotometer. OD260/OD280 ratio of ~2 and OD260/OD230 ratio of 2-2.2 were considered as an acceptable purity for the RNA samples.
The integrity and quality of the purified RNAs were further checked using 2% agarose/formaldehyde electrophoresis and ethidium bromide staining. Then 1 µg of total RNA was treated with DNase I to eliminate genomic DNA and plasmids contaminations, and subjected for the first strand cDNA synthesis using oligo dT primers and revertAid M-MulV reverse transcriptase (Thermo scientific, USA).
Real-Time Quantitative PCR (qPCR)
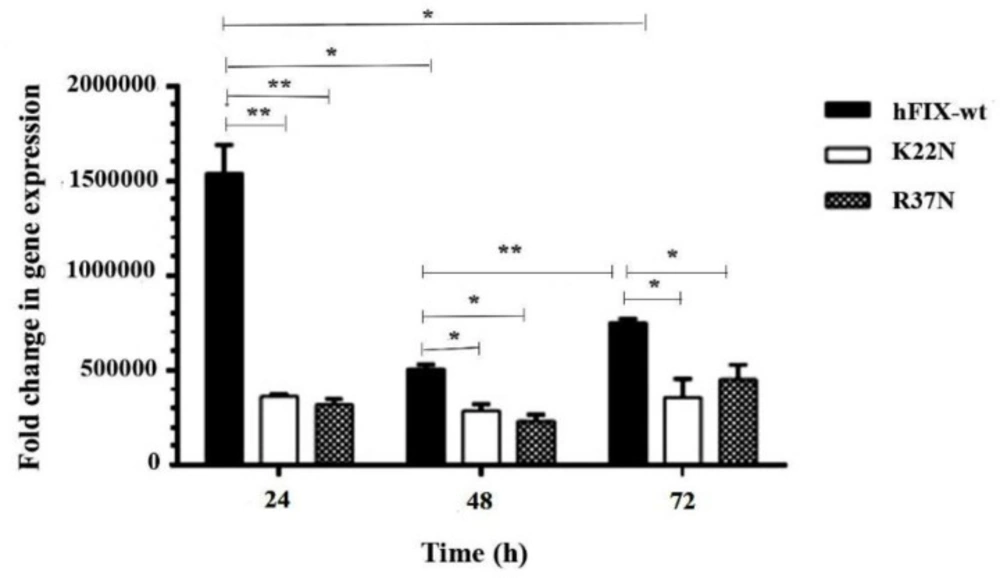
The synthesized cDNAs were then used as a template for the subsequent qPCR experiment using hFIX-specific oligonucleotide hf9-RTF1 and hf9-RTR1 as forward and reverse primers, respectively (
Table 1). A 117 bp fragment from hamster glyceraldehyde 3-phosphate dehydrogenase (Gapdh; a housekeeping gene) gene was amplified as an internal control using hams-gapdhF1 and hams-gapdhR1 primers (
Table 1). To quantify the relative mRNA level of each construct, SYBR green-based real-time PCR was carried out using RealQ Plus Master Mix Green (Ampliqon, Denmark) on Corbett Rotor-Gene 6000 Real-Time PCR machine (Qiagen, USA). The recombinant gene transcription was normalized to that of the Gapdh gene. About 50 ng cDNA sample, 0.5 μL of each forward and reverse primer (10 µM) and 5 μL RealQ Plus Master Mix Green (Ampliqon), in a final volume of 10 μL, were used for real-time PCR. The qPCR amplification temperature profile was as follow: 95 °C for 5 min (denaturation), followed by 40 cycles of amplification for 20 s at 95 °C, 20 s at 61 °C, and 20 s at 72 °C. All qPCRs were performed in triplicates. A reaction sample without cDNA was performed as a control in each run. Gapdh housekeeping gene was used as an internal control for normalization. Specificity of the primer pairs and the purity of real-time PCR products were confirmed by melting curve analysis of 65 °C to 95 °C, with fluorescence measured every 0.5°C at the end of each reaction (with no nonspecific and primer-dimer formation). Gel electrophoresis was performed to approve the PCR products and primer specificity.
Flow Cytometry Analysis
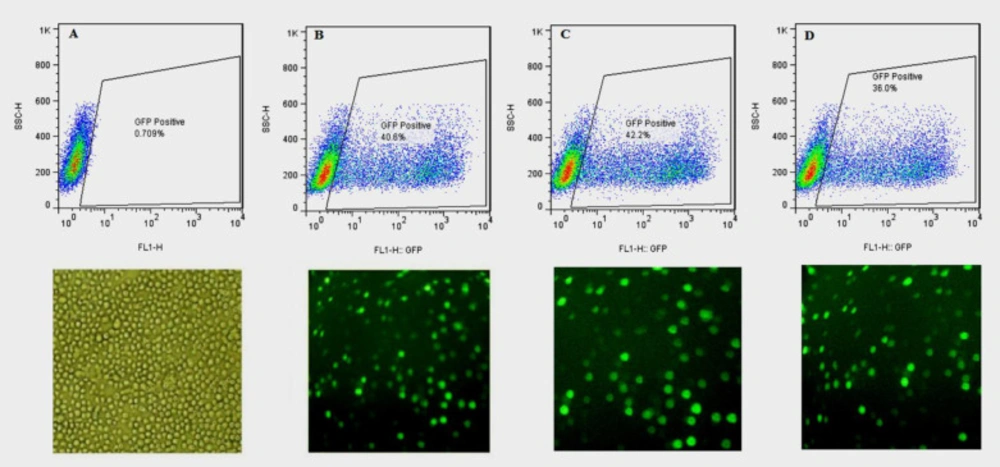
CHO-s cells transfected with bicistronic recombinant plasmids, expressing hFIXwt or its N-glycosylation mutants along with EGFP as a reporter protein, were observed using fluorescence microscope 24, 48, and 72 h after transfection to detect EGFP signals. The transfection efficiencies were then monitored using flow cytometric analysis. About 2 × 105 CHO-s cells were harvested by centrifugation at 100 ×g for 5 min, and washed twice with PBS. The cytosolic expression of EGFP in transfected cells were analyzed using flow cytometry (BD FACSCalibur, USA). EGFP fluorescence was excited at 488 nm, and emission was measured with a 530/30 nm bandpass filter. Untransfected cells were served as a control for autofluorescence and were used to set the gate for positive cells to exclude debris. The results were analyzed using FlowJo software (Treestar, Inc.).
Quantification of the Recombinant hFIX Accumulated Inside the CHO-S Cells
After 72 h of transfection, 5 × 106 CHO-s cells were pelleted by centrifugation at 100 g for 5 min. The pellets were then re-suspended in 500 μL of ice-cold lysis buffer (50 mM Tris, pH 8, 150 mM NaCl, 0.1% triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, containing protease inhibitor mix (Roche). After 20-30 min, the lysate was centrifuged at 12000 ×g for 20 min at 4 ºC, and the supernatant was analyzed for intra-cellular hFIX antigen, using ELISA.
Enzyme-Linked Immunosorbent Assay (ELISA) of hFIX Antigen (hFIX::Ag) in Culture Media
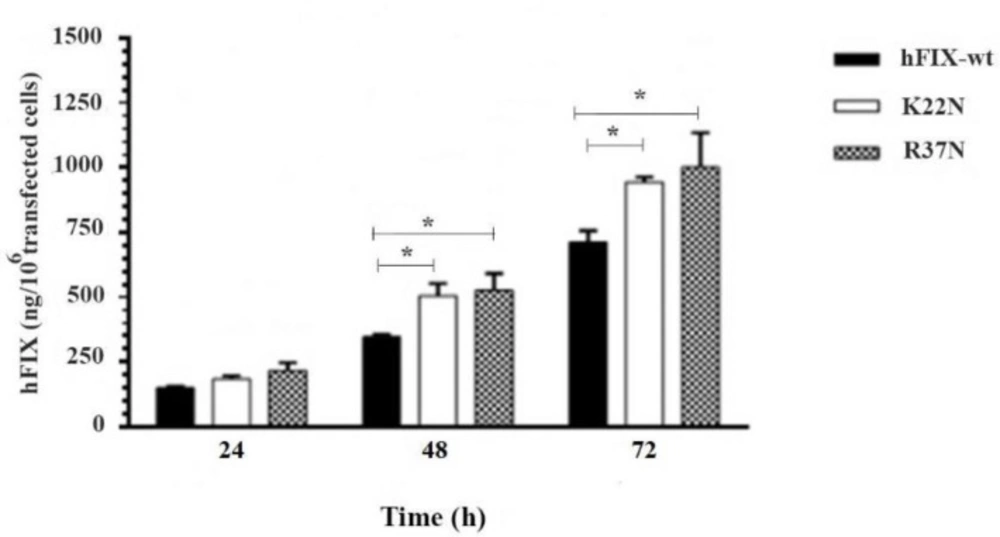
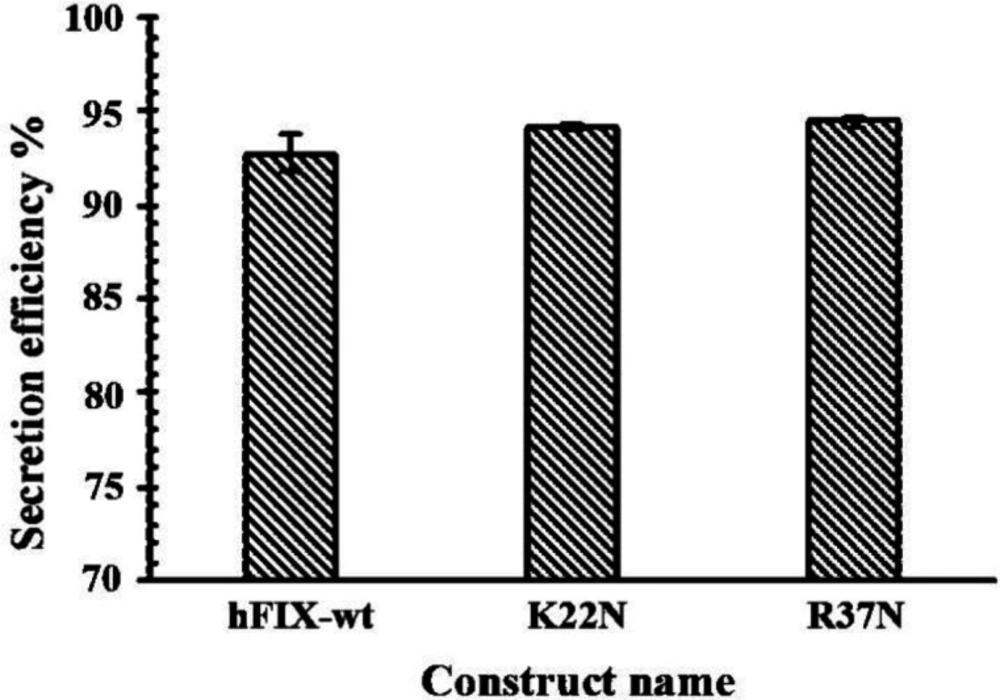
Culture media were subjected for quantification of hFIXwt or its N-glycosylation mutants using standard ELISA, based on the procedure provided by the manufacturer (Asserachrom, France). To quantify intracellular expression of recombinant hFIXwt or mutants, 50 µL of cell lysate was used for ELISA assay. The concentration of the expressed hFIXwt and mutants in culture media or intra-cell were calculated based on the standard curve and stated in ng/106 transfected cells.
Coagulation Assay
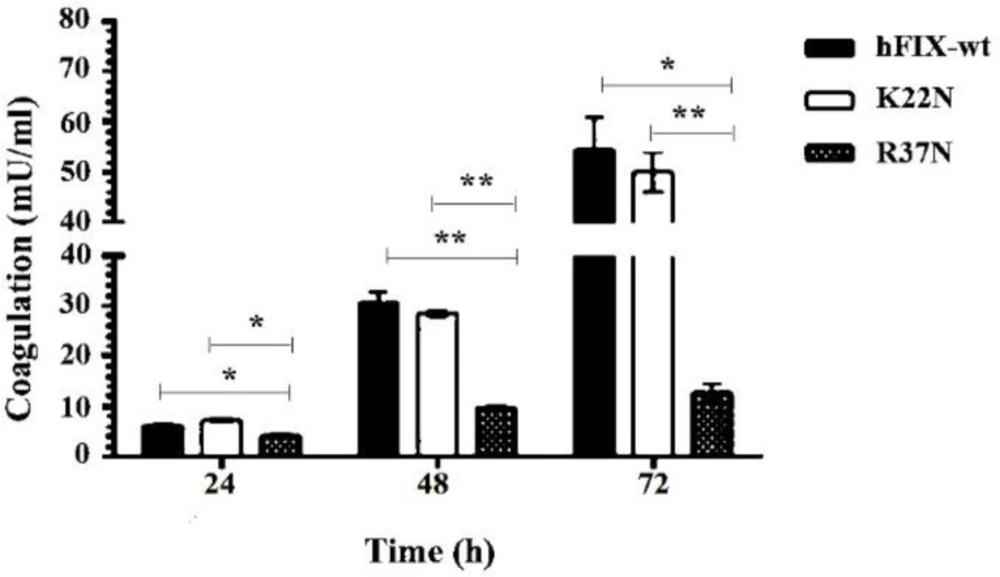
The coagulation activity of the secreted hFIXwt and its N-glycosylation mutants were measured using a chromogenic assay, according to manufacturer’s protocol (Biophen factor IX kit, France). The citrated human plasma derived from normal individuals was used as a standard sample. The culture medium from untransfected cells was used as a negative control.
The hFIX Purification
Recombinant proteins expressed by CHO-s cells transfected with bicistronic expression plasmids were then purified using Ni-NTA purification system. After 72 h of transfection, the cell suspensions were centrifuged at 100 g for 5 min and culture media were harvested. About 15 mL of the culture media was subjected for affinity purification using Ni-NTA beads (provided by Dr. Mohammadi at NIGEB, Iran).
SDS-PAGE and Western Blotting
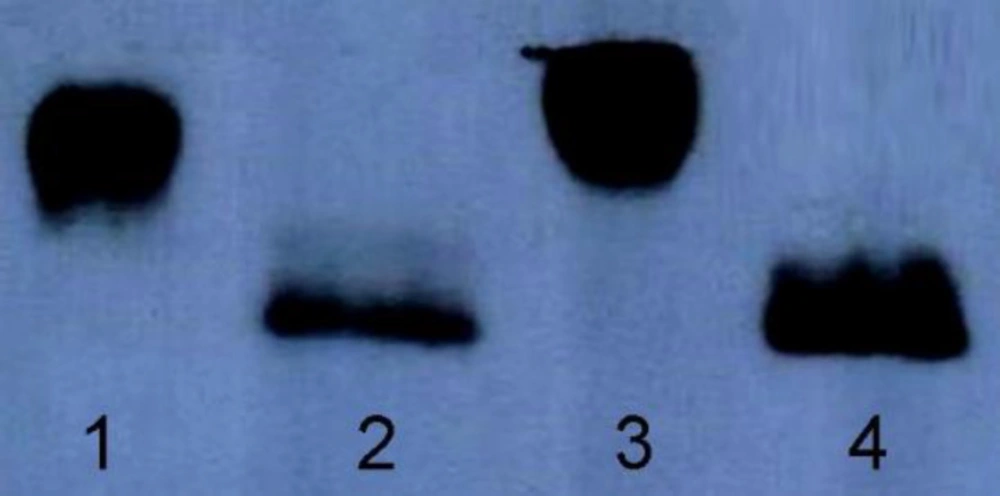
The purified recombinant hFIX and mutants were analyzed using 4–12% gradient SDS-PAGE (
31) to characterize hyper-glycosylated mutants in comparison with the native hFIX. Electroblotting of proteins onto polyvinylidene difluoride (PVDF) membrane (Roche, Germany) was performed using a wet procedure, in a transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol) for 3 h at 300 mA.
The blot was then blocked in 5% non-fat milk solution (5% w/v, in Tris-buffered Saline containing 0.1% Tween 20) and subjected for Western blotting experiment during which the electroblotted protein bands were probed with a rabbit polyclonal anti-hFIX primary antibody (Abcan, UK) with dilution of 1/2500 which was followed by incubation with goat anti-rabbit secondary antibody conjugated with peroxidase (Millipore, Co. US) with a dilution of 1/2500. Immunoreactive bands were visualized by enhanced chemiluminescence (ECL) Western blotting detection kit (Najm, Iran) or by 4-chloronaphtol substrate.
Enzymatic Deglycosylation
The purified wild-type and mutant rhFIX samples were subjected for deglycosylation using PNGase-F enzyme kit (Biolabs, New England) according to the manufacturer’s protocol. Following the denaturation step, the protein samples were treated with PNGase-F enzyme in reaction buffer containing 1% NP-40. Deglycosylated samples were then examined by 4-12% gradient SDS-PAGE and Western blotting analysis.
Statistical Analysis
All experiments were done in triplicates. The values were expressed as means±SD. The statistical analyses were performed by one-way analysis of variance (ANOVA) followed by Duncan post hoc using SPSS version 22 software. The P values less than 0.05 were considered to be statistically significant.