Preparation of the extract
Peltophorum africanum (stem bark) was selected based on our previous study. The plant was collected, identified and archived in Limpopo Province, South Africa in collaboration with a botanist at the University of Venda (voucher specimen number BP01). The extraction and the sterility test were done as previously reported (
11). Briefly, dried powder of the plant part was extracted with ethyl acetate and filtered after 48 h. The plant residue was re-extracted exhaustively and the filtrate was concentrated on a rotary evaporator (Strike 202 Steroglass, Italy) at 70
OC to take out the ethyl acetate. Working stock of the extract was prepared by sterilizing in 100% DMSO for each bioassay analysis. The extract was aseptically bottled using Acrodisc 25 mm PF Syringe (Pall, USA), tested for sterility and then stored at 4 ˚C before use. Ethyl acetate extract (EAE) was selected for this study because of its cytotoxic potency against normal human liver cell and marked antimicrobial activity as reported in our previous study (
11).
Cancer cell culture and maintenance
Cancer cell lines; MCF-7 (breast), HT-29 (colon) and HeLa (cervical) used in this study was a kind donation from Prof. Maryna Van De Venter of Nelson Mandela Metropolitan University, South Africa. Briefly, a vial containing cells (1 mL) was thawed in a water-bath regulated at 37 ˚C for 2 - 5 minutes and diluted with 9 mL pre-warmed (10 – 15 minutes in water bath at 37 ˚C) Dulbecco’s minimum essential medium (DMEM) containing 10 % fetal bovine serum (FBS). The cells were incubated in a 37 °C humidified incubator (Shel Lab, USA) at 5% CO2 for multiplication and adherence. Maintenance of cells was achieved as the old medium was aspirated, washed with 10 mL phosphate buffer saline (PBS), trypsinized (0.5 - 1 mL trypsin) and then split into a fresh medium until the desired cell number and confluence was attained.
Cell Titer-Blue viability assay
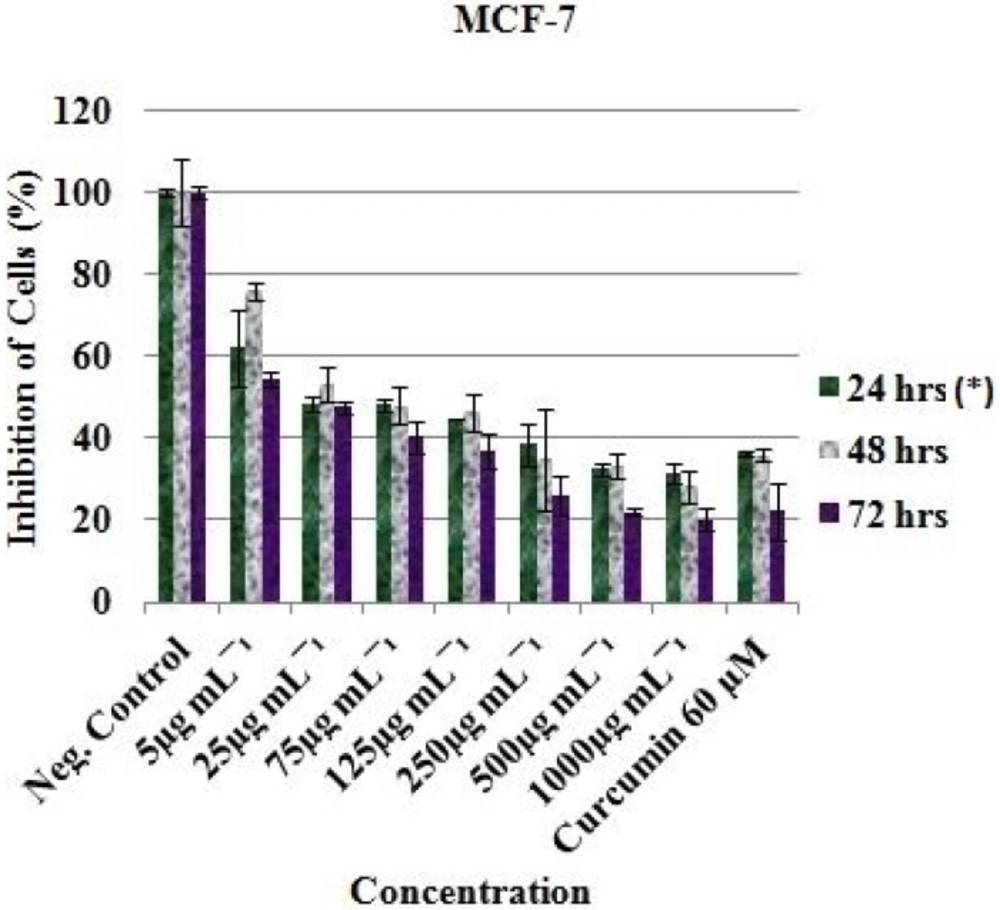
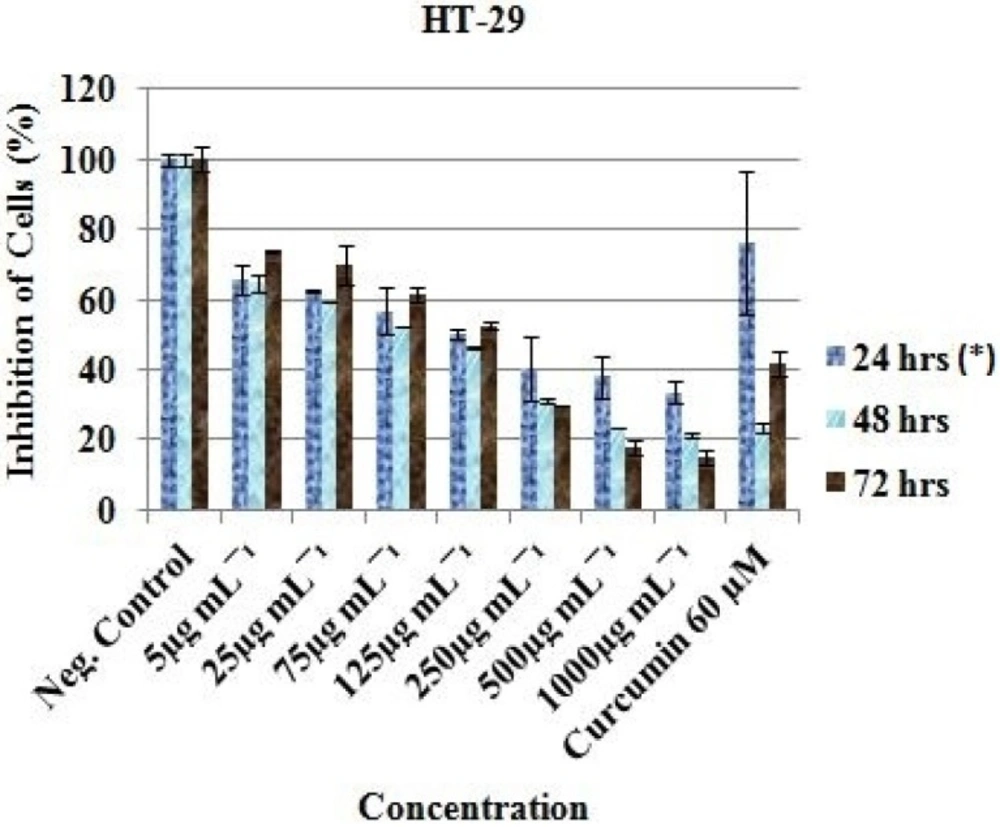
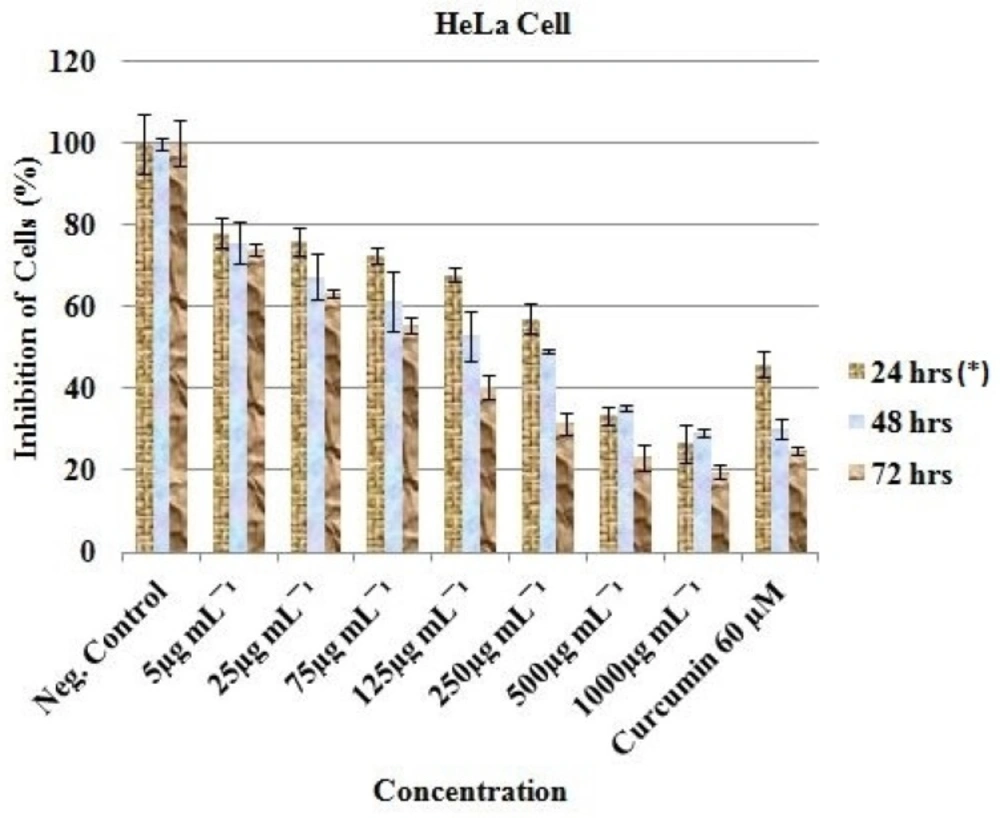
Anticancer activity of the EAE of
P. africanum was evaluated on three cancer cell lines (MCF-7, HT-29 and HeLa cell) using the microculture cell titer blue viability assay as previously described (Promega, USA) (
12,
13). The 96-well microplates were seeded with 200 μL DMEM + high glucose, L-glutamine and sodium pyruvate (Thermo Sceintific, South Logan, Utah) containing 6.0 x 10
3 cells in suspension and incubated in CO
2 (5%) incubator at 37
oC. After 24 h incubation and attachment, the cells were treated with 1000, 500, 250, 125, 75, 25 and 5 μg/mL concentration of the extract. Each plate included untreated cell controls and a blank cell-free control. Exactly 60 μM (previously reported IC
50 value) of curcumin (Sigma-Aldrich, South Africa) was used as positive control and 0.5% (residual after dilution) DMSO as negative control. After 24, 48 and 72 h of incubation, cell viability was determined by adding cell titer blue as an indicator and further incubated for 4 h. Fluorescence was read at 570/620 nm using Analytical & Diagnostic Product Gen™ spectrophotometer (BioTek, Highland Park, USA). All experiments for the extract were carried out in triplicates and the concentration which inhibited 50% of cellular growth (IC
50 value) was determined.
DNA fragmentation analysis
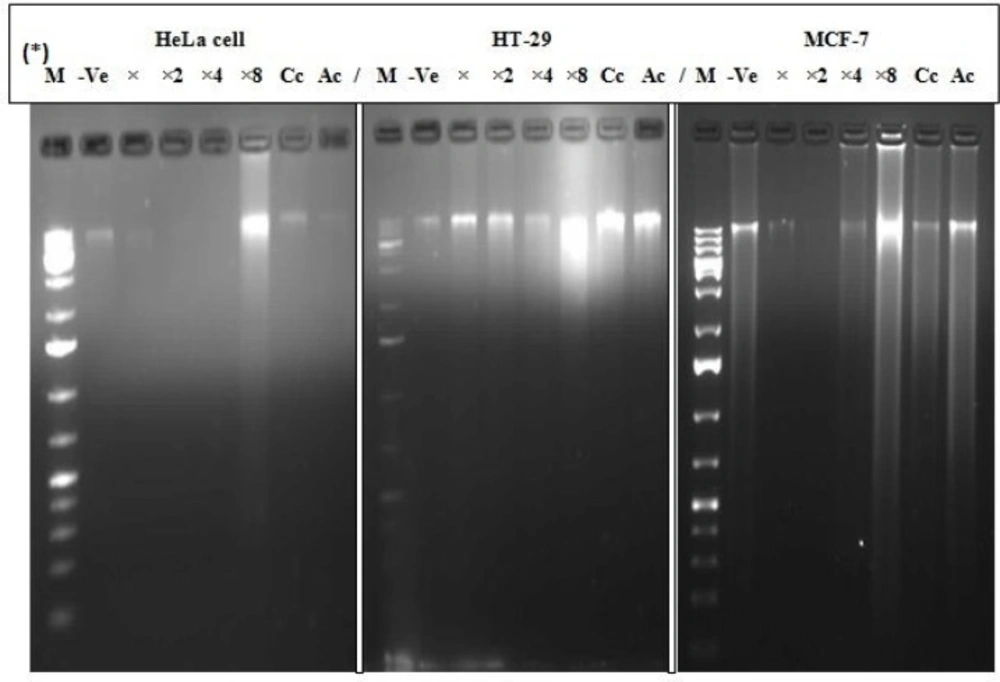
Fragmentation of the DNA was analyzed in line with a previously described method (
14). The cell lines; MCF-7, HT-29 and HeLa cell at a concentration of 1 × 10
6 mL
-1 each, were treated with EAE in a 6-well tissue culture plate after 24 h of attachment at IC
50, IC
50 × 2, IC
50 × 4 and IC
50 × 8 (in duplicate), including a negative control (untreated cells) and positive control using previously reported IC
50 value (60 µM curcumin and 10 µg/mL actinomycin D which is widely used in clinical practice since 1954 as an anticancer drug for treating many tumours with two main mechanisms of its action reported: intercalation to DNA and the stabilization of cleavable complexes of topoisomerases I and II with DNA). After 24 h of treatment, cells were harvested using sterile scraper and washed with PBS prior to DNA isolation. DNA extraction was carried out using Nucleic Acid and Protein Purification Nucleospin® Tissue Kit (Macherey-Nagel, Germany) according to the manufacturer’s manual instructions. Briefly, 10 µL of the DNA in TEA buffer was loaded onto 1.8% agarose gel containing 0.5 µg/mL (5 µL in 100 mL of gel) ethidium bromide. Electrophoresis was conducted progressively at 35, 67 and 100 V for 4 h and the DNA fragments were visualized and photographed under UV illumination XD – 79, WL/26 MX, 230 V ~ 50/60 HZ (Alliance 4.7, Taiwan, France). Based on the results obtained, we decided to proceed with analysis of only the MCF-7 cell line.
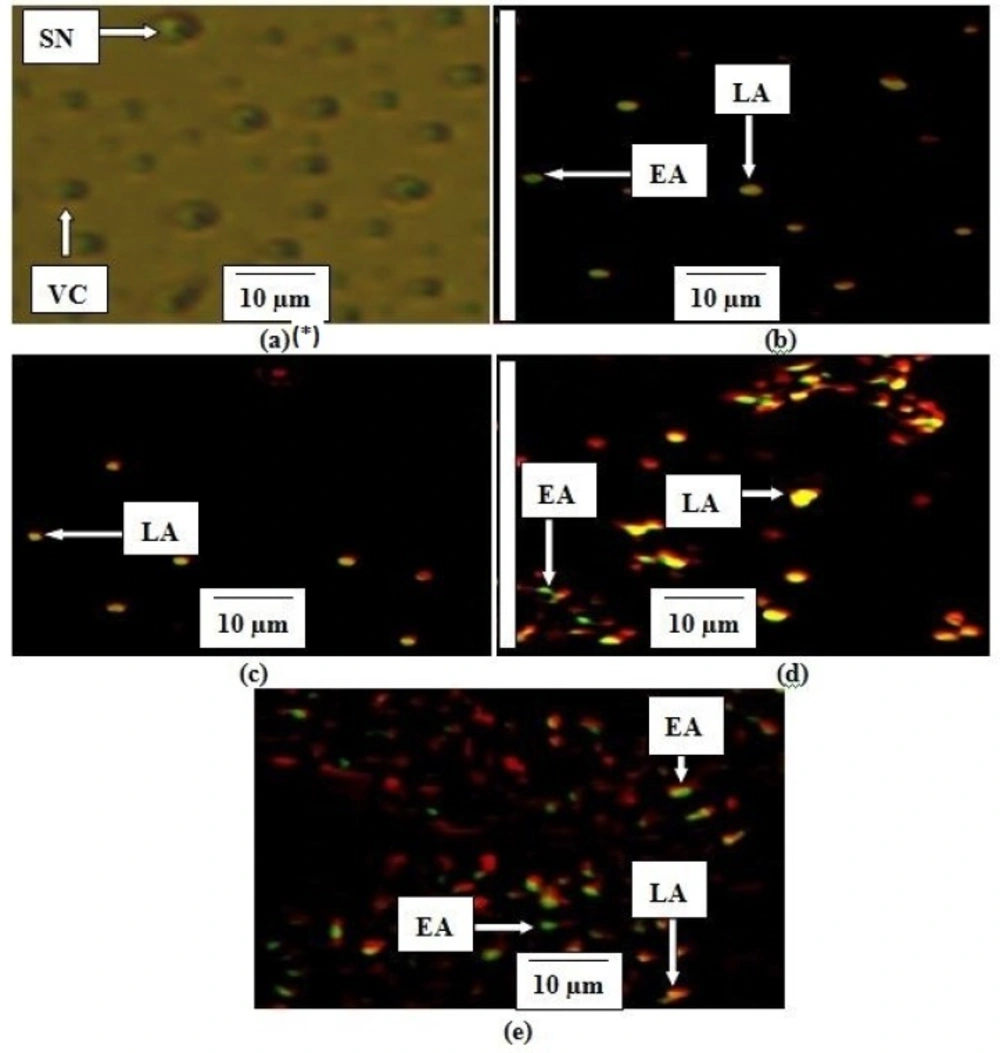
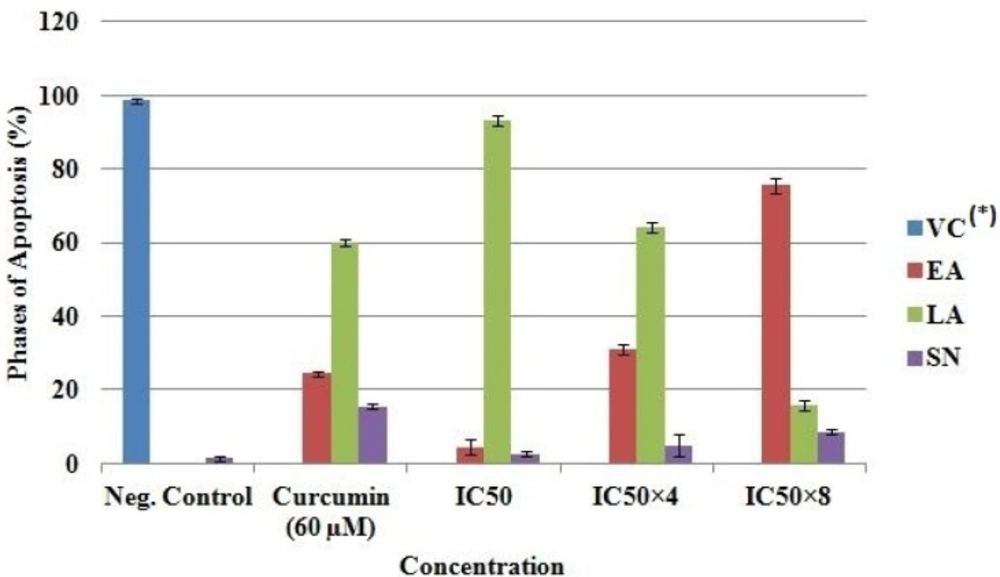
Determination of apoptosis using intercalation of nucleic acid-specific fluorochromes
Morphological changes resulting in the leakage and fragmentation of DNA by EAE were assessed under fluorescence microscope using propidium iodide (PI) and acridine orange (AO) double-staining according to the method of Mohan
et al. (
15) with few modifications. MCF-7 cells were seeded in a 6-well tissue culture plate at a concentration of 1 × 10
6 mL
-1 and treated after 24 h with EAE at IC
50, IC
50 × 4 and IC
50 × 8 respectively. Untreated cells were used as a negative control and cells treated with 60 µM of curcumin as a positive control. The experiment was performed in duplicate. The cells were collected after 24 hrs of treatment using a sterile scraper, centrifuged at 300 × g for 10 min and the cellular pellet washed twice with PBS. Fluorescent dyes; PI (10 µL) and AO (10 µL) were added to the cell (20 µL) at equal volumes and promptly observed under UV-fluorescence microscope. The percentages of viable, early apoptotic, late apoptosis and secondary necrotic cells were determined (
15).
Morphological characterization of apoptosis
The morphological characteristics of the cells (MCF-7) were determined using scanning electron microscope (SEM; JSM-6390LV, Jeol, Japan). Approximately 1 × 10
6 mL
-1 of the MCF-7 cells were seeded in a 6-well tissue culture plate and treated after 24 h with EAE at IC
50, IC
50 × 4 and IC
50 × 8. Untreated cells and cells treated with 60 µM of curcumin were included as negative and positive control respectively. Cells were collected, washed twice in phosphate buffered saline (PBS) and then centrifuged at 1000 rpm for 5 min; after which it was fixed in 2.5% gluteraldehyde prepared in 0.1 M PBS. After washing, the cells were post fixed on poly-L-lysine-coated glass coverslip, with 1 % osmium tetroxide (OsO
4) in 0.2 M PBS for 30 min, and then washed with PBS. Cells were mounted onto stubs and coated using IB3 Ion Coater (EIKO, Japan) after dehydrated through graded ethanol (30, 50, 70, 85 and 95%) and critical point dry (CPD). Different sections of the cells were micro-analyzed and the representative spectra presented (
15,
16).
Statistical analysis
Statistically significant differences among the three cell line and the apoptosis phase percentage values of MCF-7 compared with the control were determined by one way analysis of variance (ANOVA) and P. value < 0.05 was considered significant, while IC50 was determined using the regression analysis test. All analyses were performed with Minitab statistical software (student version 12 for windows).
Ethical consideration
This study which is a continuation of our line of studies on antimicrobial activity and cytotoxicity of medicinal plants had been approved by the institutional review board of the University of Fort Hare, Alice, South Africa.