Plant and chemical materials
Fresh leaves of
Avicennia alba Blume. (Acanthaceae) were collected from Lubuk Kertang village, Langkat regency, North Sumatra province, Indonesia. The samples of plant material has been identified in the Research Centre for Oceanography, Indonesian Institute of Science, Jakarta, Indonesia and the voucher specimen has been deposited in the herbarium. Roswell Park Memorial Institute-1640 (RPMI-1640) and M199 were obtained from Gibco (Carlsbad, CA, USA), the Annexin-V and propidium iodide kits were purchased from BioLegend (San Diego, CA, USA), dimethylsulfoxide (DMSO), [3-(4,5-dimetiltiazol-2-il)-2,5-difenil tetrazolium bromida] (MTT) powder, phosphate-buffered saline (PBS), and sodium dodecyl sulphate (SDS) were purchased from Sigma–Aldrich (St. Louis, MO, USA). Chloroform, methanol, hexane, ethanol, HCl, and KOH were obtained from Merck (Darmstadt, Germany). The standardization of the extract has been previously described (
13). The active polyisoprenoid compounds in the extract have been confirmed as dolichols family (C
60–C
100) (
13).
Preparation of polyisoprenoid extracts
The procedure for the extraction of polyisoprenoids was performed as previously described (
17-
19). The
A. alba leaves were dried at 60–75°C for 1–2 days. The dried leaves were crushed into a fine powder (200 g) and were immersed in 2000 mL of a mixture of chloroform:methanol (2:1, v/v) solvent for 48 h. The lipid extract of the leaves was saponified at 65°C for 24 h in 86% ethanol containing 2 M KOH. The non-saponifiable lipids of leaves were extracted with hexane, and the organic solvent was evaporated and re-dissolved in hexane.
Cytotoxic activity test
Cytotoxic activity was determined using the MTT method with slight modifications (20). Briefly, WiDr cells (1 × 10
4 cells/well) were seeded and grown in a 96-well micro plate and were incubated for 24 h. Thereafter, the cells were treated with various concentrations of PAL (500, 250, 125, 62.5, 31.25 and 15.625 µg/mL, were performed as previously described) and were incubated 37°C in a 5% CO
2 incubator for 24 h (
21). Doxorubicin was used as a positive control. DMSO (1%) was used as the co solvent. After incubation, the culture media and test solutions were discarded, and then the cells were washed with PBS. Next, 100 µL of RPMI and 10 µL of MTT (5 mg/mL) were added into each well, and the cells were incubated for 4–6 h in a 5% CO
2 incubator at a temperature of 37°C. The MTT reaction was stopped using a stopping reagent (10% SDS in 0.1 N HCl), and the cells were protected from light at room temperature and allowed to stand for one night. Cell viability was observed using an ELISA reader (BioTek EL 800, United States) at a wavelength of 595 nm. Living cells react with MTT to form a purple colour. The percentage viability was calculated using the following formula (
22):
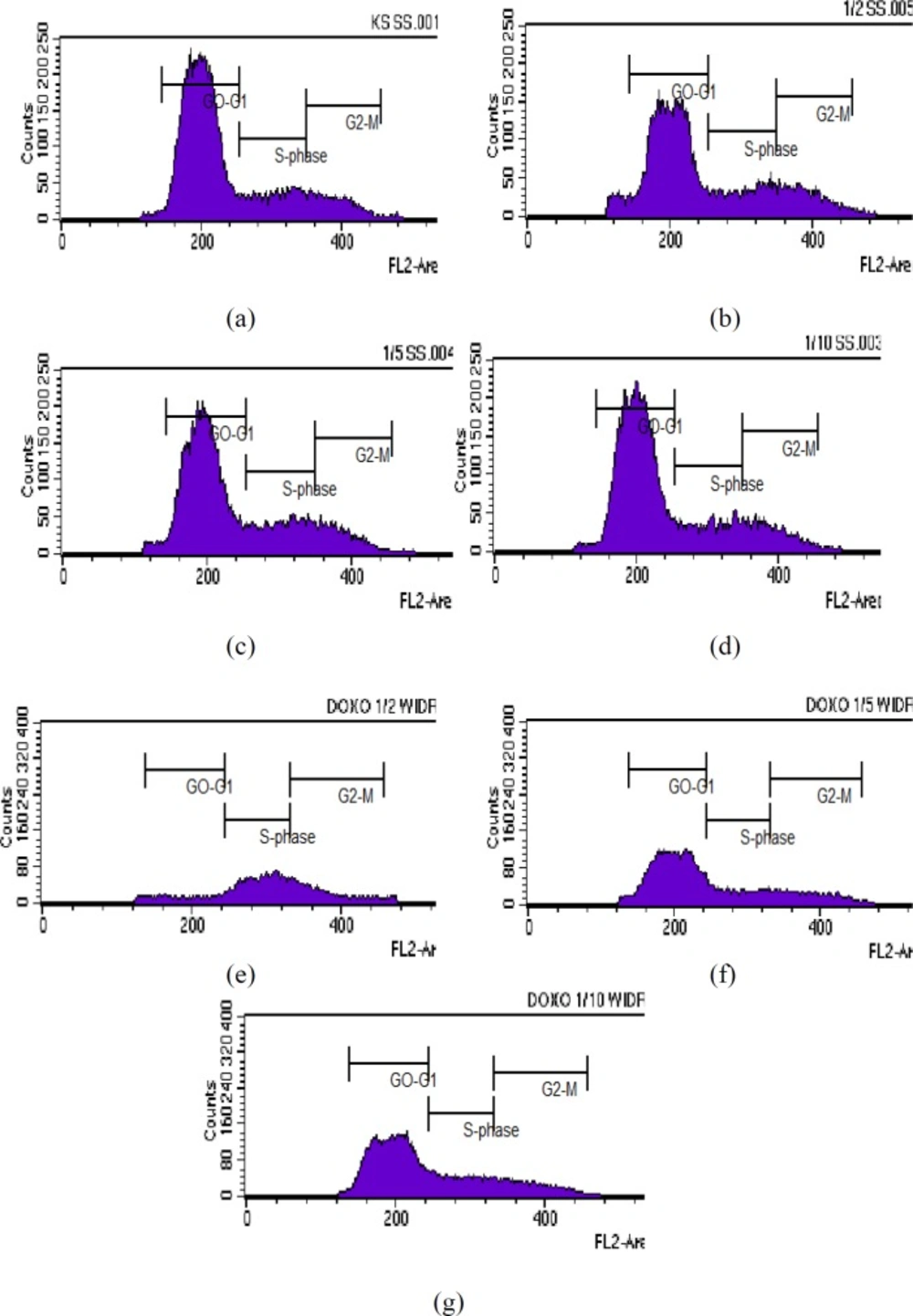
Cell cycle inhibition analysis
Cell cycle inhibition was assessed as described previously (
23). Briefly, WiDr cells were platedin a 6-well microplate at adensity of approximately 5 × 10
5 cells/well and then were incubated for 24 h to obtain good growth. The next day, the cells were treated with various concentrations of PAL (1/2, 1/5 and 1/10 IC
50, were carried out as previously reported) and were incubated for 24 h (
21). Doxorubicin was used as a positive control. After incubation, the samples were transferred into 15-mL conical tubes, and the microplates were washed with PBS, which was then collected and added to the same conical tubes. Next, 250 µL of trypsin was added to the microplates, which were then incubated for 3 min at 37°C. Thereafter, 1 mL of culture media was added to the microplates, and then the media were collected and added to the same conical tubes. Next, 1 mL of PBS was added to the microplates, and then the PBS washes were collected and added to the same conical tubes, followed by centrifugation at 600 rpm for 5 min, and removal of the supernatants. Thereafter, the pellets were resuspended in 1 mL of PBS, followed by transfer to microtubes and centrifugation at 2000 rpm for 3 min. Next, 500 µL of 70% ethanol was added and incubated for 30 min. After incubation, the cells were resuspended in propidium iodide. The cell cycle distribution was observed using a flow cytometer (FACSCalibur).
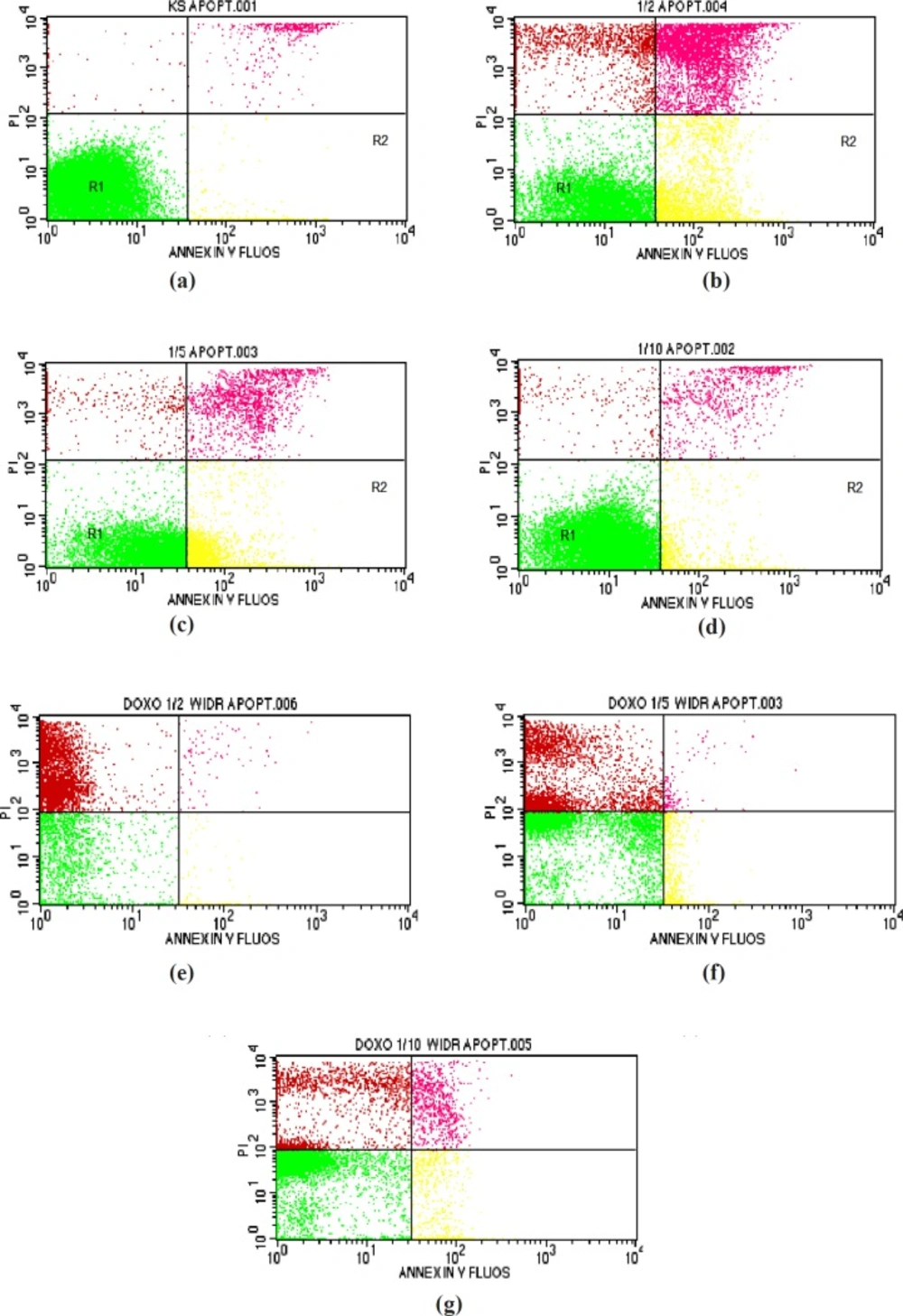
Apoptosis analysis
Apoptosis detection was performed as described previously (
23). Briefly, WiDr cells were grown in a 6-well microplate at a density of approximately 5 × 10
5 cells/wells and incubated for 24 h to obtain good growth. The following day, the cells were treated with various concentrations of PAL (1/2, 1/5, and 1/10 IC
50) and incubated for 24 h. Doxorubicin was used as a positive control. After incubation, the samples were transferred into 15-mL conical tubes, the microplates were washed with PBS, and then the PBS washes were collected and added to the same conical tubes. Next, 250 µL of trypsin was added to the microplates, which were incubated for 3 min at 37°C. Thereafter, 1 mL of culture media was added to the microplates, and then the media were collected and added to the same conical tubes. Next, the cells in the microplates were resuspended in 1 mL PBS, the PBS washes were collected and added to the same conical tubes, followed by centrifugation at 600 rpm for 5 min and removal of the supernatants. The pellets were dissolved in 1 mL of PBS and were transferred into microtubes and centrifuged at 2000 rpm for 3 min. Next, 100 µL of Annexin-V buffer was added to the cells, followed by the addition of 5 µL each of Annexin-V and propidium iodide and incubation for 10 min. After incubation, 300 µL of Annexin-V buffer was added. Apoptosis was observed using a flow cytometer (FACSCalibur).
Suppression of COX-2 expression
Suppression of COX-2 was performed as described previously (
24). Briefly, WiDr cells were grown on coverslips in24-well microplate at a density of 5 × 10
4 cells/well and were incubated for 24 h to obtain good growth. The following day, the cells were treated with various concentrations of PAL (1/2, 1/5, and 1/10 IC
50) and were incubated for 24 h. Doxorubicin was used as a positive control (
25). After incubation, the cells were washed with PBS. Next, cold methanol was added to the microplates, which were incubated in the freezer at –4°C for 10 min. After incubation, the methanol was removed, and the coverslips were placed on a dish. Next, the cells were washed with distilled water three times and were incubated with hydrogen peroxide blocking solution for 10 min at room temperature, followed by removal of the blocking solution. Next, the cells were incubated with prediluted blocking serum for 10 min at room temperature, followed by removal of the serum. After incubation, the COX-2 antibody was added, followed by incubation for 1 h at room temperature. After incubation, the coverslips were washed with PBS and then were incubated with secondary antibody (biotinylated universal secondary antibody) for 10 min. After incubation, the cells were washed with PBS and then were incubated with streptavidin-horse radish peroxidase enzyme and incubated for 10 min. Next, the cells were washed with PBS, DAB was added, and then the cells were incubated for 5 min (for brown colour development). After incubation, the cells were washed with PBS and distilled water, and then the cells were washed with Mayer-Haematoxylin solution and were incubated for 5 min. After incubation, the cells were washed with distilled water, followed by the addition of 70% ethanol and incubation for 2 min. Next, xylol solution was added to the cells, followed by drying. After drying, mounting media was added dropwise to the coverslips, which were covered with slides. Suppression of COX-2 was observed using a microscope with the optiLab system.
Statistical analysis
Data were represented as means ± SD from at least three independent experiments. The IC50 values was calculated from the linear regression equations of dose response curve for each experiment using probit analysis with SPSS 23 software.