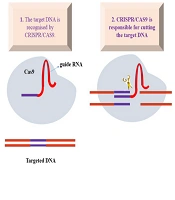
In this study, we successfully established a human cellular model of CDKL5-related ASD using CRISPR/Cas9-mediated genome editing. By introducing a specific missense mutation (c.172A>T; p.Lys58Met) within exon 2 of CDKL5, we generated a controlled and reproducible platform for investigating the molecular pathology of CDKL5 deficiency. This model recapitulated key hallmarks of the disorder, including downregulation of CDKL5 expression, compromised cell viability, and increased apoptosis, thereby validating its utility for mechanistic and therapeutic research. The CRISPR/Cas9 system enabled efficient and specific gene editing, with an average on-target activity of 41.6% and an HDR efficiency of 12%. Importantly, no detectable off-target mutations were identified at the top predicted loci, confirming the specificity of the sgRNA design.
These results are consistent with recent reports using CRISPR/Cas9 to manipulate CDKL5 in neuronal and glial cells. Carriero et al. used CRISPR to model CDKL5 deficiency in rat hippocampal neurons, demonstrating impaired synaptic vesicle endocytosis and altered neuronal excitability. Similarly, Halmai et al. used a dCas9-TET1 fusion to epigenetically reactivate CDKL5 on the inactive X chromosome (
24). Collectively, these findings and the present results demonstrate that CRISPR systems, whether nuclease-active or catalytically inactive, can recapitulate CDKL5 dysfunction and provide a foundation for therapeutic correction studies.
The observed 55% reduction in CDKL5 mRNA and 48% reduction in protein expression in edited clones suggest that the p.Lys58Met substitution impairs transcript stability and/or translational efficiency. This amino acid lies within the conserved N-terminal serine/threonine kinase domain, a critical region required for substrate phosphorylation and catalytic activity (
4,
5). Previous studies have shown that missense mutations in the kinase domain often destabilize the protein or disrupt its subcellular localization (
6). Reduced CDKL5 expression has profound implications for neuronal development. In animal models, Cdkl5 knockout leads to dendritic spine immaturity, defective synaptic plasticity, and behavioral deficits reminiscent of ASD (
7,
8). The present cellular findings support these observations and confirm that even a single amino acid substitution within the catalytic region can trigger downstream molecular dysfunction.
The increased apoptosis observed in the mutant clones, characterized by a 2.3-fold elevation in caspase-3 activity and a higher proportion of Annexin V-positive cells, corroborates previous evidence that CDKL5 contributes to neuronal survival. Loi et al. demonstrated that Cdkl5-deficient neurons exhibit heightened susceptibility to excitotoxic and oxidative stress, likely due to a defective DNA damage response (
25). Similarly, Nicole et al. (2021) reported mitochondrial dysfunction and bioenergetic deficits in neurons derived from CDKL5-mutant iPSC lines (
26). In the present study, cell death occurred independently of Cas9 toxicity, as control cells transfected with the empty PX458 vector maintained normal viability. These results suggest that CDKL5 deficiency directly activates apoptotic cascades, possibly through mitochondrial membrane depolarization or impaired phosphorylation of neuroprotective targets, such as MeCP2 and HDAC4 (
11,
12). Indeed, CDKL5-mediated phosphorylation of MeCP2 is essential for chromatin regulation and synaptic maturation, and disruption of this pathway may account for overlapping phenotypes between CDKL5 deficiency and Rett syndrome (
13).
Several previous studies have used patient-derived fibroblasts or iPSCs to study CDKL5 deficiency; however, such systems often exhibit high genetic variability. For instance, Nicole et al. (2021) showed that CDKL5 mutations in iPSC-derived neurons cause mitochondrial transport defects and abnormal calcium homeostasis (
26). However, these lines inherently contain patient-specific polymorphisms, making it difficult to distinguish primary disease-causing effects from secondary, background-related effects. The generation of a CDKL5-mutant human cell model provides an important platform for understanding the molecular relationship between ASD and epileptic encephalopathies. CDKL5 interacts with numerous ASD-related genes, including SHANK3, NRXN1, and MECP2, converging on pathways involved in synaptic regulation and chromatin remodeling (
15). Disruption of these networks contributes to an abnormal excitatory/inhibitory balance in cortical circuits, a hallmark of autism (
16,
17).
CDKL5 phosphorylates key synaptic proteins, including MeCP2 at Ser421 and MAP1S, thereby modulating chromatin architecture and microtubule stability, respectively. The p.Lys58Met mutation, located in the ATP-binding pocket of the kinase domain, is predicted to abolish catalytic activity, leading to hypophosphorylation of these substrates. This aligns with the observed reduction in cell viability and increase in apoptosis, phenotypes also reported in SHANK3 R1117X knock-in models and SCN2A L1342P iPSC-derived neurons, in which single-nucleotide substitutions similarly induced caspase-3 activation and metabolic stress without overt neuronal differentiation (
27).
Although CDD is primarily associated with impaired neuronal development and function, HEK293T cells were intentionally selected in this study as an initial human cellular platform for CRISPR/Cas9-mediated knock-in generation rather than as a complete neuronal disease model. HEK293T cells offer several technical advantages for precise genome editing, including high transfection efficiency, rapid and robust proliferation, reproducible culture conditions, and efficient clonal expansion and screening after editing. These features are particularly important for knock-in strategies, in which HDR-mediated precise editing is technically challenging and often requires extensive screening and validation of edited clones. In this context, the HEK293T-based system enabled reliable introduction and validation of the pathogenic CDKL5 c.172A>T variant and allowed assessment of its immediate molecular and cellular consequences, including reduced CDKL5 expression, decreased cell viability, increased apoptosis, and evaluation of predicted off-target loci.
However, the non-neuronal origin of HEK293T cells limits their ability to model neuron-specific aspects of CDD, such as synaptic maturation, dendritic development, calcium signaling, and electrophysiological activity. Therefore, this model should be interpreted as an initial molecular validation platform, and future studies using iPSC-derived neurons, neuronal precursor cells, or other disease-relevant neuronal systems will be necessary to extend these findings in a more physiologically relevant context. While this study demonstrates the successful generation of a CDKL5-mutant cellular model, certain limitations should be acknowledged. First, HEK293T cells are non-neuronal and may not fully capture the neuron-specific regulatory dynamics of CDKL5. Future studies using neuronal precursor cells or iPSC-derived neurons would provide improved physiological relevance. Second, the analysis focused on gene expression and apoptosis; however, additional functional assays, such as electrophysiological recordings, calcium imaging, or synaptic-marker quantification, could further elucidate the impact of CDKL5 loss on neuronal connectivity. Finally, although off-target analysis revealed no significant mutations in the top 5 predicted sites, comprehensive genome-wide off-target mapping using GUIDE-seq or whole-genome sequencing would enhance confidence in the genetic precision of the model.
5.1. Conclusions
This study successfully established a human cellular model of ASD associated with CDKL5 deficiency using the CRISPR/Cas9 genome-editing system. By introducing a defined missense mutation (c.172A>T; p.Lys58Met) into exon 2 of CDKL5, we achieved precise genomic modification with high specificity and reproducibility. The resulting mutant cells displayed markedly reduced CDKL5 mRNA and protein levels, decreased cell viability, and significantly increased apoptosis. These findings validate CDKL5 as a central regulator of cellular survival pathways and highlight its essential role in neuronal integrity. This model provides a useful in vitro platform for exploring the molecular underpinnings of CDKL5-related neurodevelopmental disorders and testing emerging therapeutic strategies, such as CRISPR-based correction, epigenetic reactivation, and small-molecule modulation. Although the HEK293T-based model offers a useful and reproducible platform for preliminary mechanistic studies and therapeutic screening, its non-neuronal origin limits its ability to fully recapitulate the neuronal context of CDD. Accordingly, future studies using iPSC-derived neuronal models or neuronal cell lines will be necessary to validate and extend these findings in more physiologically relevant contexts.




 Colony PCR analysis showing the expected approximately 230-bp amplicon in representative transformed colonies. L, DNA ladder; N, negative control; S1-S6, representative positive colonies. Overall, 23 of 25 colonies were positive, indicating a cloning efficiency of 92%. (B) Sanger sequencing chromatogram confirming correct sgRNA insertion into the PX458 vector; the boxed region represents the 20-nt sgRNA target sequence. Results are representative of 3 independent biological replicates.")
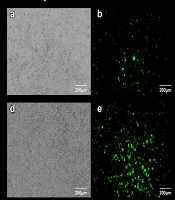
, eGFP fluorescence (B), and merged (C) images of HEK293T cells at 18 hours after transfection. (D-F) Representative bright-field (D), eGFP fluorescence (E), and merged (F) images of HEK293T cells at 36 hours after transfection. Scale bars: 200 μm. All images are representative of 3 independent biological replicates (n = 3).")
 and the mutant-specific reaction (M24), indicating the presence of both wild-type and c.172A>T mutant alleles and confirming successful heterozygous HDR-mediated editing. B, Representative Sanger sequencing chromatogram verifying the c.172A>T substitution, which results in the p.Lys58Met amino acid change. Data are representative of 3 independent biological replicates (n = 3).")
. Statistical significance was determined using an unpaired 2-tailed Student <i>t</i>-test. **P < 0.01.")
, with each experiment performed in technical triplicate. Statistical significance: * P < 0.05 and ** P < 0.01 compared with wild-type controls.")
 mutant HEK293T cells. A, Representative Western blot images showing GAPDH (approximately 37 kDa) as a loading control. B, Representative Western blot images of CDKL5 (approximately 115 kDa) in wild-type (WT) and mutant (MUT) cells across 3 independent biological replicates. C, Densitometric quantification of CDKL5 protein levels normalized to GAPDH. Data are presented as mean ± SD (n = 3). CDKL5 expression was significantly reduced in mutant cells (0.65 ± 0.01) compared with WT controls (1.24 ± 0.03), representing an approximately 48% decrease (*** P < 0.001, unpaired Student <i>t</i>-test).")