Preparation of nanoparticles containing Hydroxyurea
In order to prepare nanoparticles containing Hydroxyurea , 1.99 g (0.01 mol) of FeCl2.4H2O and 5.41 g (0.02 mol) of FeCl3.6H2O were dissolved in 50 mL of distilled water and polyethylene glycol was added to the solution. Then ammonium hydroxide solution was added drop-by-drop until saturation. The reaction temperature was set on 80 ºC. Then, a homogenizer stirred the solution for 60 min at a speed of 10,000 rpm. After the reaction, the obtained black sediment was washed with distilled water three times and then dried under vacuum. The obtained solution was sonicated using a sonicator (Bandelin Sonorex Digitec) for 5 min to uniformly distribute nanoparticles inside the solution; 15 mg of Hydroxyurea drug was added to 5 mL of suspension of pegylated iron oxide nanoparticles (0.5 mg/mL). Then it was incubated for 48 h at room temperature on a magnetic stirrer at the speed of 600 rpm.
Analysis of nanoparticle and nanoparticle-loaded Hydroxyurea characteristics
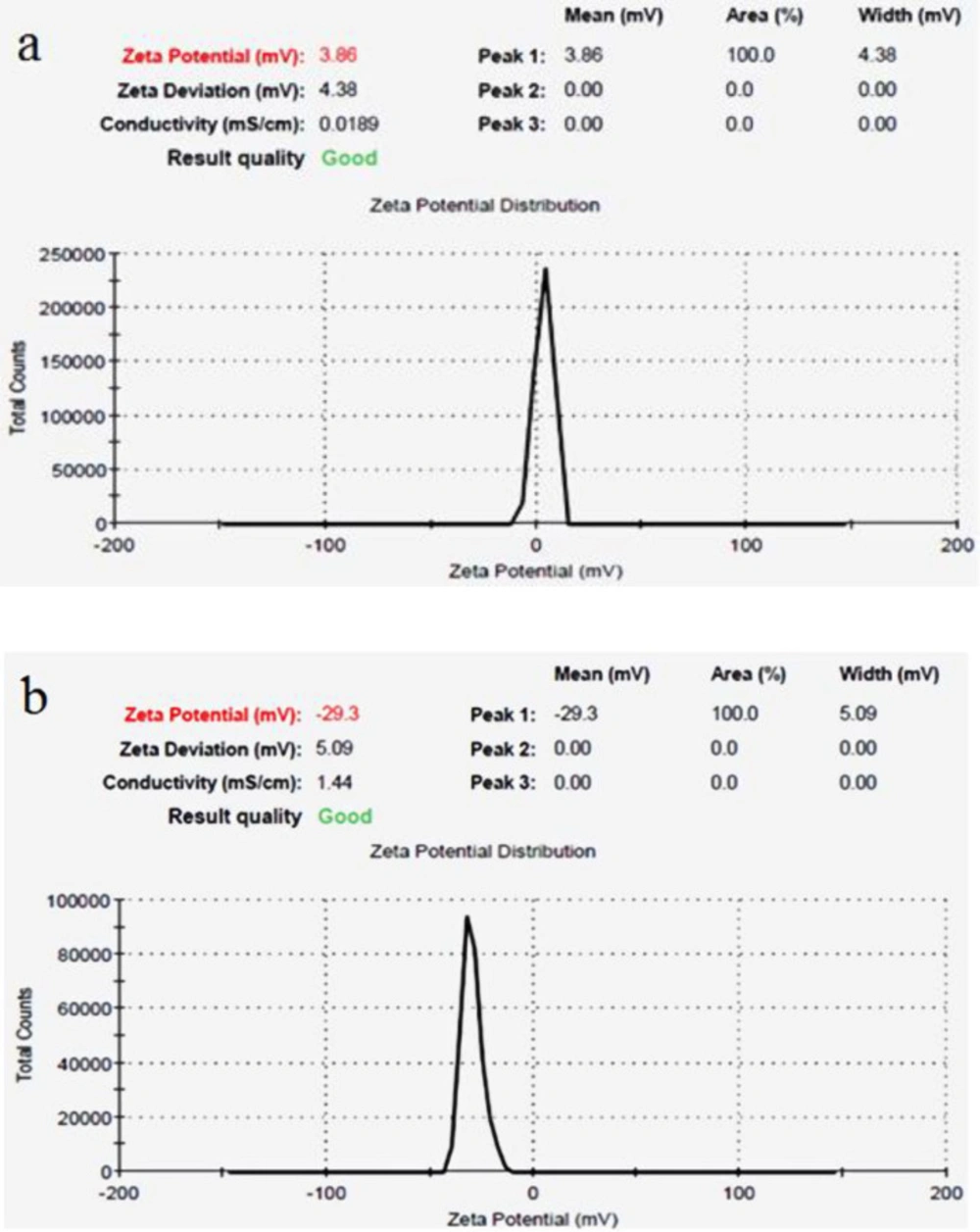
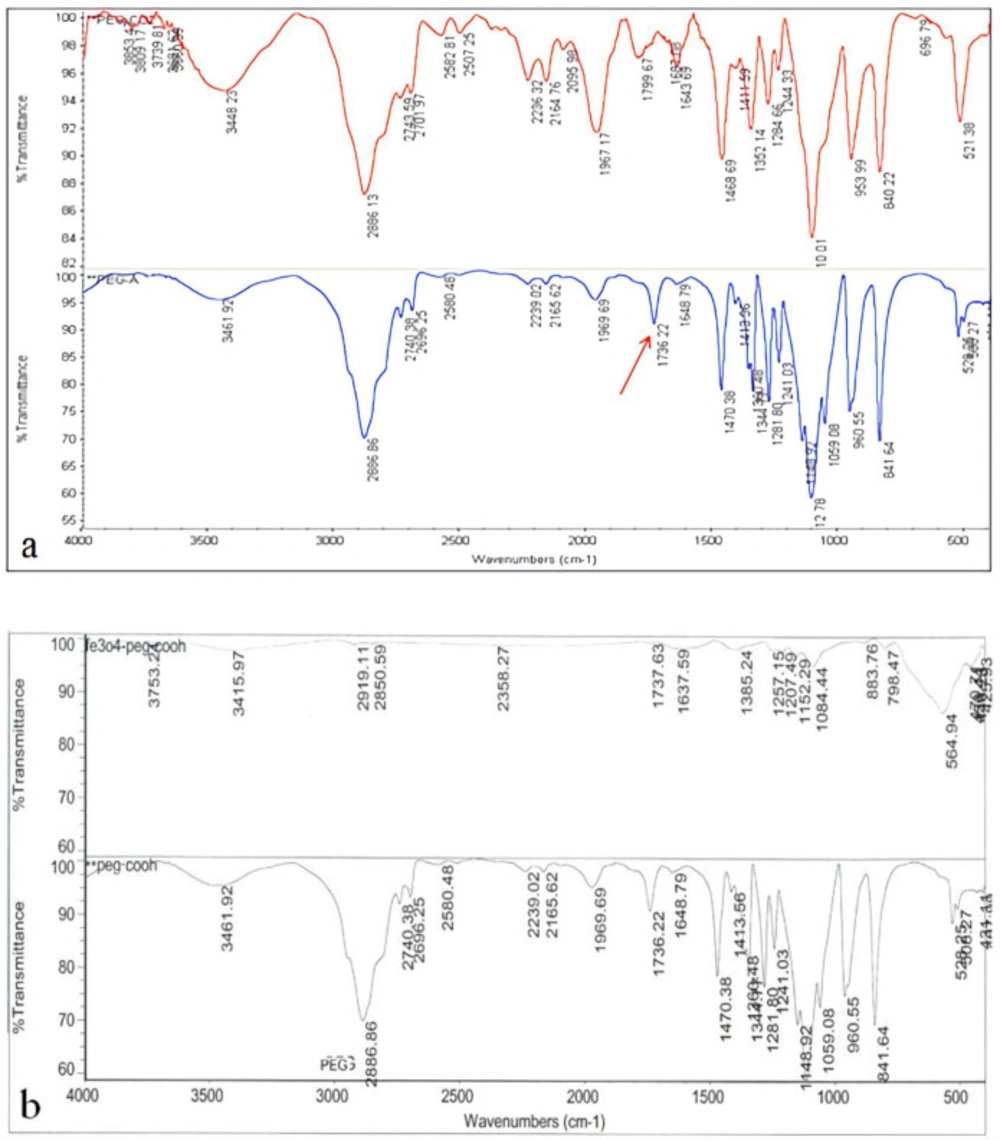
Size and morphological characteristics of synthesized nanoparticles were assessed using the scanning electron microscope. The surface potential of magnetic iron oxide nanoparticles was assessed by Malvern Zetasizer 3000 HSA. Fourier transform infrared spectroscopy (FTIR) technique was used to ensure the formation of iron oxide nanoparticles bond with the polyethylene glycol polymer and the conversionof polyethylene glycol to acidic polyethylene glycol. Finally, Hydroxyurea loading on pegylated nanoparticles was assessed by FTIR technique.
Cell culture
The cells were cultured based on methods described by others (Elengoe and Hamdan 2013). In brief, MCF-7 breast carcinoma cells (ATCC, Manassas, VA) were cultured in DMEM/F12, 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (Gibco, Carlsbad, CA). The Cells were cultured as adherent monolayers (i.e., cultured at approximately 80% confluence) and maintained at 37 °C in a humidified atmosphere of 5% CO2. The cells were harvested with 0.25% trypsin and 1 mM EDTA (Gibco, USA). All chemicals used were of research grade.
Study design
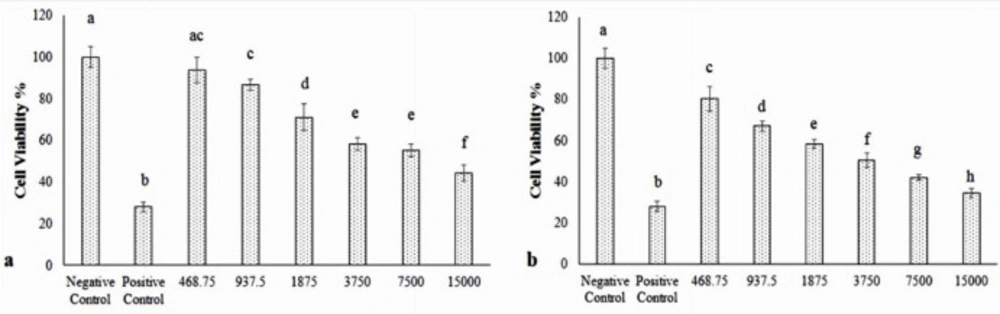
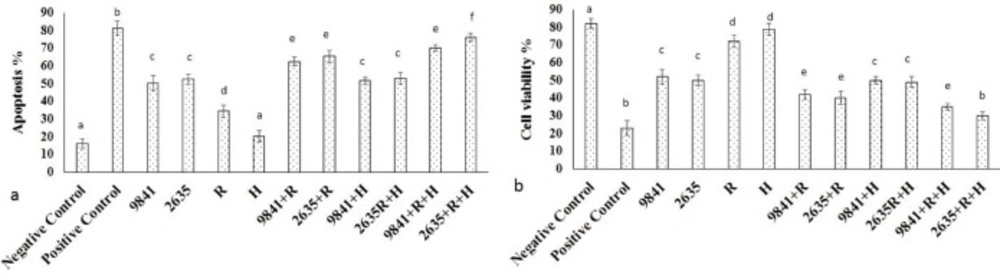
The cultured MCF-7 cells were randomized into T25 flasks and divided into 14 groups. Group 1 was negative control. Group 2 treated with Doxorubicin (800 nM) as the positive control. Group 3-8 treated with the different doses of Hydroxyurea (468.75, 937.5, 1875, 3750, 7500, and 15000 μM). Group 9-14 receiving the different doses of nanoparticle-Loaded Hydroxyurea (468.75, 937.5, 1875, 3750, 7500, and 15000 μM). In the third stage, MCF-7 treated with IC50 of Hydroxyurea and nanoparticle-loaded Hydroxyurea in combination with radiation and hyperthermia. After 24 h of incubation, proliferation of MCF-7 cell was evaluated.
Hyperthermic chemosensitization experiments
In chemosensitization experiments, the cells were plated in a 96-well plate in the appropriate culture medium. The cells, simultaneously with the drug treatment, were incubated for 1 h at 41 ºC.
Irradiation
The cells were irradiated with a single dose from a cobalt-60Co radiation source (gamma radiation). In the experiment, the cells were exposed to a dose of 2Gy with 1.25 MeV.
Measurement of apoptosis
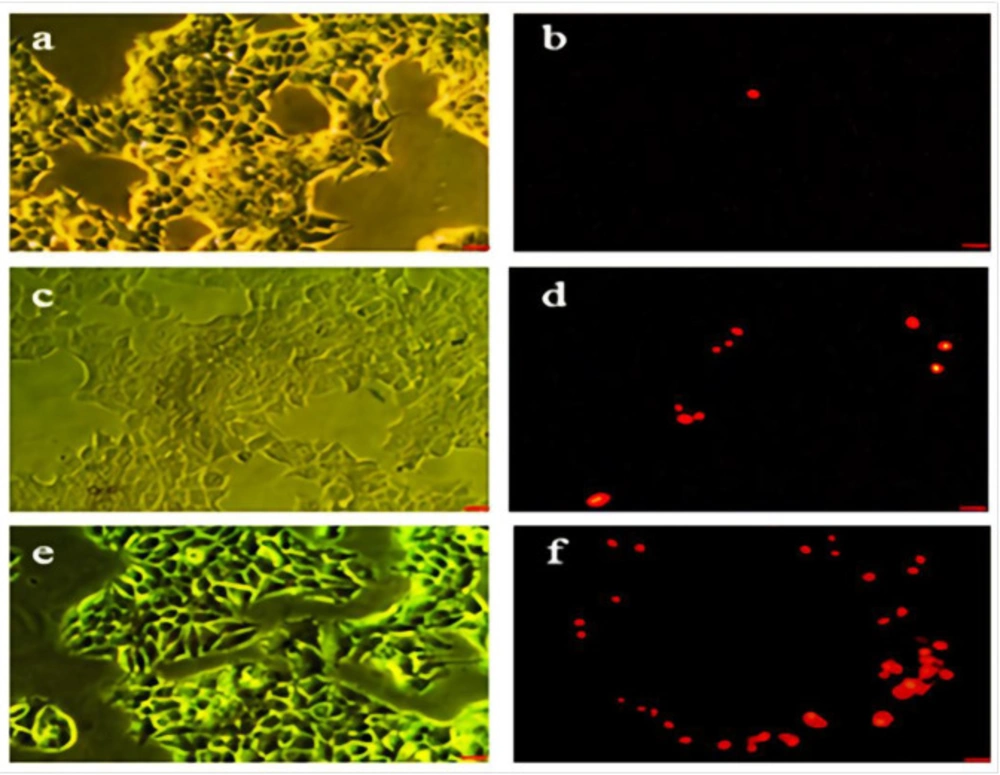
Acridine orange (AO) and propidium iodide (PI) were used to visualize living and dead cell for estimating the viability of the cells. The MCF-7 cells were incubated with Hydroxyurea and nanoparticle-loaded Hydroxyurea for 24 h. One group treated with Doxorubicin (800 nM) as positive control. They were then stained with 10 µL/mL acridine orange and incubated for 15 min in 37 °C and Co2 5%. The cells were washed twice with PBS and stained with 10 µL propidium iodide. The cells were then incubated for 5 min in 37 °C and Co2 5%. The cells were again washed with PBS and centrifuged in 1400 rpm for 10 min. The supernatant was removed and 50 µL of the pellet was mounted on the glass slide and observed under fluorescent microscopy. The 100 cells were counted in 5 field and apoptotic and live cells were determined. Acridine orange and propidium iodide are nucleic acid binding dyes that can be used to measure the cell viability. Since acridine orange is cell permeable, all stained nucleated cells generategreen fluorescence. Propidium iodide only enters the cells with compromised membranes and therefore dying, dead, and necrotic nucleated cells stained with propidium iodide generate a red fluorescence.
Acridine orange and propidium iodide are nuclear staining dyes. AO is permeable to both live and dead cells and stains all nucleated cells to generate green fluorescence. PI enters dead cells with compromised membranes and stains all dead nucleated cells to generate red fluorescence (
16).
Measurement of cell viability
The cultured cells were harvested, counted (10000 cells per well), and transferred to 96-well plates and incubated for 24 h prior to the addition of appropriate treatments. The treated cells were incubated for 24 h. MTT (3-(4, 5-dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide) (5 mg) was dissolved in 1 mL of phosphate-buffered saline (PBS), and 25 μL of the MTT solution was added to each of the 96-well plate. The plates were wrapped in aluminum foil and incubated at 37 °C for 4 h. The solution in each well, containing media, unbound MTT, and dead cells, was removed by suction, then 200 μL of DMSO was added to each well. The plates were then shaken, and the optical density was measured using a micro plate reader at 492 nm. Three independent experiments were performed for each study and all measurements were performed in triplicate. The results were expressed as the percentage proliferation with respect to vehicle-treated cells. The growth inhibition of at least 50% was considered as cytotoxic. One group was treated with Doxorubicin (800 nM) as the positive control (
17).
Measurement of Caspase-8 and 9 Activities
A quantitative enzymatic activity assay was carried out according to the instructions of the manufacturer for the colorimetric assay kit. After treatment with various concentrations of Hydroxyurea, nanoparticle-loaded Hydroxyurea and Doxorubicin in 800 nM, cells were washed, collected, lysed, centrifuged, and analyzed for total protein by the Bradford assay. Twenty microlitre of supernatants was added immediately to a buffer containing a p-itroaniline (pNA)-conjugated substrate for caspase-8 (Ac-IETD-pNA) and (LEHD-pNlabeled) for caspase 9. The samples were incubated for 1 h at 37 °C. Absorbance was measured at 405 nm in a plate reader (
18).
Statistical Analysis
Experimental results were expressed as means ± SD. Statistical analyses were performed using PASW 18.0 (SPSS Inc., Chicago, IL, USA). Model assumptions were evaluated by examining the residual plot. Results were analyzed using a factorial ANOVA with two between-subjects factors. Bonferroni test for pairwise comparisons was used. The differences were considered significant when P < 0.05.