Materials
Dulbecco’s Modified Eagle’s Medium (DMEM), Ham’s F12 medium (F12 nutrient medium), and Fetal Bovine Serum (FBS) were purchased from Gibco (USA). Penicillin, streptomycin, L-glutamine, and TMB (3,3ʹ,5,5)ʹ-tetramethylbenzidine) substrate were obtained from Invitrogen (USA). The Ph.D.TM-7 phage display peptide library kit was purchased from New England BioLabs (MA, USA).IPTG (isopropyl β-D-thiogalactoside) and X-gal (5-bromo-4-chloro-3-indolylβ-D-galactoside) were obtained from Appli Chem (Germany). Taq DNA polymerase 2X Master Mix Red was purchased from Amplicon (Denmark). GeneAll® ExpinTM GelSV Kit was purchased from GeneAll Biotechnology Co. (South Korea). Mouse anti-M13 phage antibody and horse radish peroxidase (HRP)-conjugated rabbit anti-mouse secondary antibody were purchased from Abcam Inc (MA, USA). All chemicals, buffers, and bacterial culture media were obtained from Merck (USA).
Cell culture
Human lung adenocarcinoma cell line A549, human hepatocellular carcinoma cell line Huh-7, human esophageal squamous cell carcinoma cell line KYSE-30, human breast adenocarcinoma cell line MCF-7, human normal lung epithelial cells SAEC, and human normal fibroblast cells were used in the study. All cell lines were purchased from Cell Bank of Pasteur Institute (Tehran, Iran) except for SAEC that was a gift from Zanjan University. Tumor cell lines were maintained in DMEM supplemented with 10% FBS, penicillin (100 U/mL), streptomycin (100 mg/mL), and 2 mM L-glutamine. Normal fibroblast cells -used for the depletion of the randomized peptide library– and normal lung epithelial cells were grown in a medium containing 1:1 mixture of DMEM and Ham’s F12 medium supplemented with 10% FBS, growth factors, and penicillin/streptomycin antibiotics. All cells were grown in 25 cm2 polystyrene culture flasks or six-well culture plates until they reached sub-confluent monolayers. The cells in culture were maintained at 37 °C in a humidified atmosphere of 95% air and 5% CO2. For routine maintenance, cells were passaged by trypsinization before becoming fully confluent. None of the cell types were retained in continuous culture for more than 1 month.
Phage display library
The Ph.D.TM-7 phage display peptide library kit contains random seven amino acid peptides fused in-frame to the N-terminus of the minor coat protein (pIII) of the filamentous M13KE phage. Therefore, 3-5 copies of a foreign peptide are expressed as part of the minor coat protein on the surface of each phage particle. The library titer is 1×1013pfu/mL (plaque forming units). The library has a complexity of 1×109 individual phage clones. This level of complexity represents approximately all possible 7-mer peptide sequences that can be expressed by a random heptapeptide sequence. Extensive sequencing has revealed the presence of a huge diversity of displayed peptide variants with no considerable positional bias in the naïve library. The Escherichia coli strain ER2738 which is a robust F+ strain with a rapid growth rate and particularly well-suited for M13KE amplification was used for phage propagation.
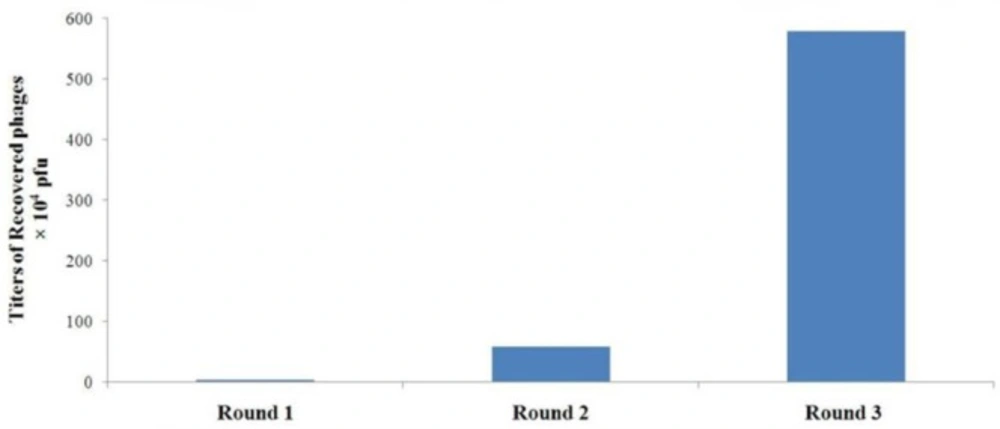
| Fold Increase | Recovery Efficiency | Output Number (pfu) | Input Number (pfu) | Round of Panning |
|---|
| 17.21 | 3.4 × 10-7 | 3.4 × 104 | 1 × 1011 | 1 |
| 9.88 | 5.85 × 10-6 | 5.85 × 105 | 1 × 1011 | 2 |
| 170.03 | 5.782 × 10-5 | 5.782 × 106 | 1 × 1011 | 3 |
| Frequency Percent | Frequency | Peptide Sequence | Peptide Name | Phage Name | Phage Clone |
|---|
| 42 | 5 | AWRTHTP | LCP1 | P1 | PC1, PC3, PC7, PC8, PC11 |
| 17 | 2 | THSNLSV | LCP2 | P2 | PC2, PC10 |
| 17 | 2 | AFRDPLY | LCP3 | P3 | PC4, PC6 |
| 8 | 1 | THLSVNK | LCP4 | P4 | PC5 |
| 8 | 1 | NGAYRAI | LCP5 | P5 | PC9 |
| 8 | 1 | LEQTPMF | LCP6 | P6 | PC12 |
| Sequence | Peptide Name |
|---|
| V | S | L | N | S | H | T | LCP2 |
| K | N | V | S | L | H | T | LCP4 |
| F | M | P | T | Q | E | L | LCP6 |
| Y | L | P | D | R | F | A | LCP3 |
| P | T | H | T | R | W | A | LCP1 |
| I | A | R | Y | A | G | N | LCP5 |
Specific enrichment of A549 cell-binding phages during rounds of in-vitro panning The titers of the recovered phages from each round were determined by blue plaque-forming assay on IPTG-Xgal agar plates containing tetracycline
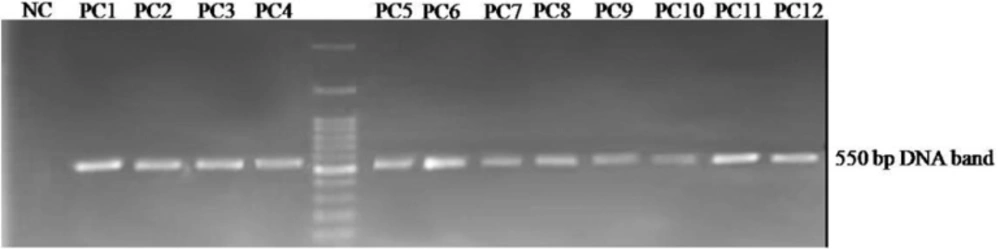
Agarose gel electrophoresis of the displayed peptide-encoding DNA insert obtained from selected phagesAn approximately 550 bp fragment is amplified by PCR on the isolated phage genomes. PC1 to PC12: Phage plaques selected from the third round of panning, NC: negative control.
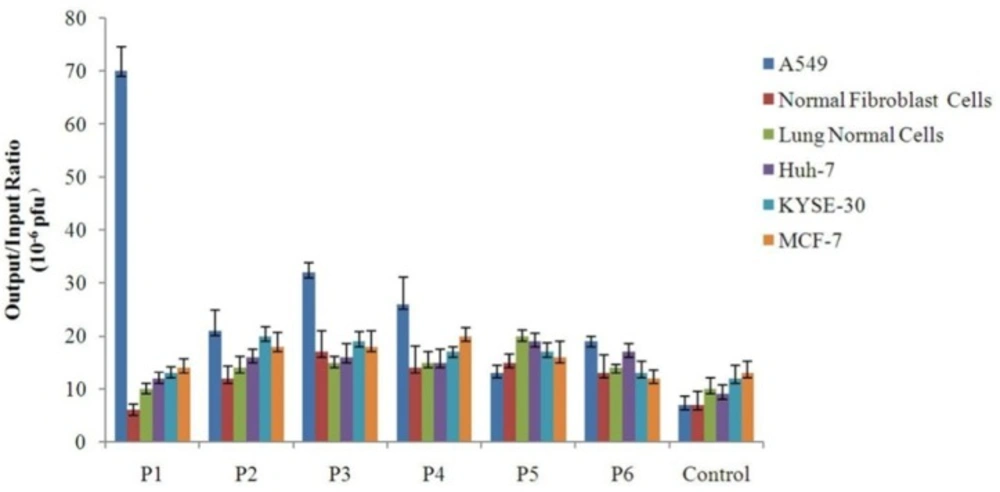
Binding of the selected phage clones to different cell types Each cell was grown to sub-confluent monolayer and then incubated individually with 109 pfu of different selected phage clones. The unbound phages were washed away. Afterwards, the cell-bound phages were collected by using a low-pH buffer, and quantitated through infection of bacterial cells and titering by blue plaque-forming assay. Binding efficiency of each clone to each cell type was determined by calculating the output/input ratio. A phage with an unrelated displayed peptide was used as control for cell binding assay
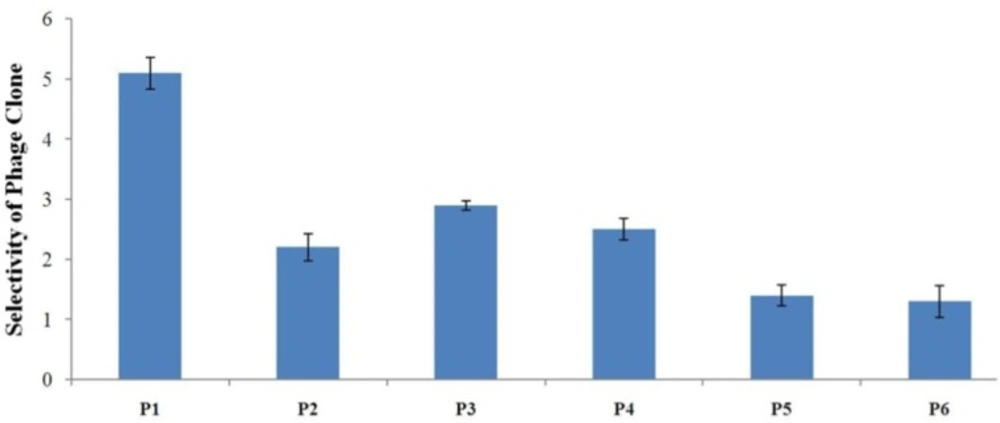
Evaluation of the binding selectivity of the selected phage clones by cell ELISA Cells were seeded onto 96-well cell culture plates overnight (1×104 cells/well). 109 phage particles were added to each well. Phage binding to cells was detected through adding mouse anti-M13 phage antibody and HRP-conjugated rabbit anti-mouse secondary antibody. OD was obtained after blocking the reaction. The selectivity values for phage binding was calculated by the formula mentioned in the text and were 5.1, 2.2, 2.9, 2.5, 1.4, and 1.3 for P1, P2, P3, P4, P5, and P6, respectively. P1 is indicated to be the strongest binder and binds more effectively than all phage clones to A549 cells
In-vitro screening of phage peptide library
A modified subtraction and selection protocol was optimized and used for the screening of the Ph.D.
TM-7 phage display peptide library (
18,
19). A549 was taken as the target cell for positive selection and normal fibroblast and Huh-7 were taken as the absorber or control cells for subtractive panning. Briefly, cells were seeded onto six-well plates and cultured in DMEM medium containing 10% FBS at 37 °C in a humidified atmosphere with 5% CO
2.In the first round of panning, a rigorous library depletion procedure was performed in which the naïve library was successively subjected to empty multi-well plates, serum, and absorber cells (fibroblast and Huh-7 cells) before being incubated with target A549 cells. To do this depletion plan, an aliquot of the library mixed with blocking buffer was serially incubated with depleting agents for 1 h at 37 °C. Blocking buffer consisted of 2% BSA in phosphate buffered saline (PBS). In each step of the depletion screening, supernatant was removed from the well and used for the following depletion step. In the second and third rounds of selection, only control cells were exploited for subtractive panning.
During rounds of panning, cells were washed with PBS and kept in serum-free medium for 1 h at 37 °C. Before phage application, the cells were blocked with blocking buffer for 1 h. Then, an aliquot of the library that contained 1011pfu was added to control cells and incubated for 1 h with gentle shaking. The non-binding phages were removed and added to target A549 cells.After incubation for 1 h, the unbound or weakly-bound phages were wiped off by extensive washing three times with PBS and five times with tris buffered saline containing tween 20 (TBST). The cell surface-bound phages were recovered by treating A549 cells with elution buffer (0.2 M glycine/HCl, pH = 2.2) for 10 min on ice and then neutralizing with 1M Tris-HCl. A small amount of the recovered phages was used for titering through blue plaque-forming assay on LB agar plates containing IPTG and X-gal. The remaining phages were amplified by infecting with ER2738 bacterial culture to be used as input for the next round of in-vitro selection. During rounds of selection, panning intensity was progressively enhanced by increasing the number of washing times with PBS and TBST from 8 for the first round to 12 for the last round.
Amplification of phage clones
ER2738 strain was inoculated into LB medium to achieve log-phase bacterial culture. The phage suspension recovered from each round of panning was added to 20-25 mL of LB medium containing ER2738 and the mixture was incubated at 37 °C with shaking for 4.5-5 h. Following precipitation of bacterial cells by centrifugation, 1/6 volume of 20% polyethylene glycol (PEG)/2.5M NaCl was added to the recovered supernatant and incubated at 4 °C overnight or on ice for 2 h. After two successive PEG/NaCl precipitation steps, phage particles were harvested by centrifugation at 13000 rpm for 20 min. The resulting phage pellet was resuspended in tris buffered saline (TBS).
Phage titering
To determine the phage titer, phages were serially diluted in liquid LB and each diluted solution was incubated with a log-phase culture of ER2738 for 5-10 min at room temperature to allow infection of bacterial cells by phage particles. 2-3 mL of melted top agar (45 °C) was added and the infected bacteria were spread on LB agar plates containing IPTG (50mg/mL) and Xgal (40 mg/mL). Plates were incubated overnight at 37 °C. Finally, the titer of the phage solution was calculated by counting the number of blue plaques appeared on the plate after 12-16 h and using the following formula:
Where a is the number of blue plaques, b is the times of dilution, and d is the plated volume of the phage suspension (μL)
PCR amplification and DNA sequencing of selected phages
Plaque PCR was performed to amplify the inserts encoding peptides displayed on the surface of the selected phages. A portion of an individual well-isolated plaque was scraped up by using a pipette tip and then dipped into a PCR tube containing 25 μL of Taq DNA polymerase 2X master mix red, 2.5 μL of each of the forward and reverse primers (20 mM), and 20 μLof deionized water to reach the total reaction volume of 50 μL. Taq DNA polymerase 2X master mix red consists of the following components: 0.2 units/μL of Taq DNA polymerase, the NH4+ buffer system, 0.4 mM of each dNTP, and 1.5 mM MgCl2. The sequences of the forward and reverse primers were as follows: Forward primer:5ʹ-TTTAGTCCTCAAAGCCTCTG-3ʹ and Reverse primer: 5ʹ-CAAGCCCAATAGGAACCC- 3ʹ. The primers were designed by using Oligo Primer Analysis Software v.7 The phage DNA was amplified by using the following PCR program in a thermal cycler: 30 cycles (30 sec at 94 °C, 30 sec at 60 °C, and 30 sec at 72 °C) preceded by an initial denaturation for 5 min at 95 °C and followed by a final extension for 10 min at 72 °C. The amplified DNA was purified from gel by using GeneAll®ExpinTMGelSV Kit and finally sequenced by ABI 3700 (Applied Biosystems, USA).
In-vitro cellular binding efficiency of phage clones
A549, Huh-7, KYSE-30, MCF-7, normal lung epithelial cells, and normal fibroblasts were used for cellular binding assays. Briefly, 1×104 cells were grown in each well of a 96-well cell culture plate for 24h to reach sub-confluent monolayers. Cells were then incubated with serum-free medium for 1h.1×109pfu of each phage clone was added separately to different cell types and incubated for 1h at 37 °C with gentle agitation every 5 min. The unbound phages were carefully removed from the wells and the cells were washed 6-8 times with cold washing buffer (3-5 min for each time). Cell-associated phages were collected by adding 100 μL of an acidic elution buffer for 10-15 min on ice and then neutralizing with 100 μL of 1M Tris-HCl. The retrieved cell-bound phages were titered by infecting bacteria and phage binding efficiency was calculated through dividing the number of output (recovered) phages by the number of input phages. Phage displaying an unrelated peptide was used as control.
Cell ELISA with phage
Cells were cultured in DMEM and seeded onto 96-well plates (1×10
4 cells/well) the day before use. Cells were then incubated with serum-free medium at 37 °C for 1h and fixed by using ice-cold 4% paraformaldehyde (PFA) for 10-15 min. The fixed cells were washed with PBST (PBS containing 0.05% w/v Tween 20) and blocked with 100-200 μL of blocking buffer at 37 °C for 1.5-2 h. Phage clones were added into wells (1×10
9pfu/well) and the plates were kept at 37 °C for 1h. Subsequently, the unbound phages were removed by washing three times with TBST. To detect the bound phages, cells were incubated at 37 °C for 1h with 100 μL/well of mouse anti-M13 phage antibody, washed three times with PBST, and then incubated with 100 μL/well of HRP-conjugated rabbit anti-mouse secondary antibody. After washing, color development was carried out by adding 100 μL of freshly prepared TMB substrate to each well and incubating the plate in the dark for 5 min. The reaction was terminated via adding 50-100 μL of 0.5 M H
2SO
4 and the absorbance (OD) values were measured by an automated ELISA plate reader (BioTek Instruments Inc, USA) at wavelength of 450 nm. Selectivity of phage clones was calculated by using the following formula (
20);
whereS1 and C1 represent the OD values obtained from binding of the selected phage and control phage to A549 cells and S2 and C2 represent the OD values obtained from binding of the selected phage and control phage to control cells.
Statistical analysis
All statistical analyses for in-vitro cellular binding assay and cell ELISA were performed by using Microsoft Office Excel 2007 and Graph Pad Prism 5. Statistical differences among samples were evaluated by one-way ANOVA and P< 0.05 was considered as statistically significant.