4.1. Compositional and Positional Analyses
A comparison of AAC between hiABFs and QSPs was conducted in this study, the results of which are shown in
Figure 3A. It can be observed that hiABFs exhibited a higher abundance of positively charged amino acids, such as arginine (R) and lysine (K), as well as amino acids with hydrophobic side chains, including leucine (L), tryptophan (W), and valine (V), compared to QSPs. Appendix 4 in the supplementary file provides a classification of amino acids, based on their side chain properties. Twenty standard amino acids were categorized into five groups based on their properties. The aliphatic group was composed of G, A, V, L, M, and I amino acids, while the aromatic group consisted of F, Y, and W amino acids. The positive charge group included K, R, and H amino acids, while the negative charge group comprised D and E amino acids. Also, the uncharged group included S, T, C, P, N, and Q amino acids. The distribution of these groups between hiABFs and QSPs is presented in
Figure 3B.
A, comparison of amino acid composition (AAC) between highly active antibiofilms (hiABFs) and quorum-sensing peptides (QSPs); B, the figure demonstrates the distribution of aliphatic, aromatic, positively charged, negatively charged, and uncharged amino acids in hiABFs and QSPs.
The bacterial membrane contained a significant number of negatively charged components, which made it favorable for interactions with positively charged peptides through cation-pi interactions. In this regard, Segev-Zarko et al. demonstrated that a high frequency of leucine (L) and lysine (K) in peptide sequences could enhance their ABF effects (
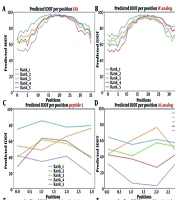
51). Positional preference analyses were conducted for a comprehensive assessment of peptide sequences, exhibiting high ABF activity. The analysis focused on the first five positions of both N and C termini. Sequences with a length exceeding 10 residues were selected from hiABF and QSP datasets.
Figure 4A and
B depict the results of positional preference analysis.
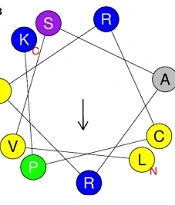
The sequence logos of the first five positions of N and C termini in A, highly active antibiofilms (hiABFs); and B, quorum-sensing peptides (QSPs). The size of residues is proportional to their propensity.
In hiABFs, the N-terminal positions were predominantly occupied by arginine (R) and lysine (K), followed by hydrophobic amino acids, including leucine (L) and isoleucine (I). In QSPs, the first position was predominantly occupied by the uncharged and polar amino acid, serine (S), along with negatively charged amino acids, including aspartate (D) and glutamate (E). Serine also exhibited dominance in the third and fifth positions, while glycine (G) and leucine (L) were more frequently observed in the second and fourth positions.
In the C-terminus of hiABFs, arginine (R) and lysine (K) were the most preferred residues in all five positions. In contrast, in QSPs, the first to fourth positions were predominantly occupied by nonpolar amino acids, including phenylalanine (F), alanine (A), leucine (L), and glycine (G). In the fifth position, positively charged residue lysine (K) was predominant. In this regard, a study by Rydberg et al. focused on three peptide sequences, which were exclusively composed of arginine (R) and tryptophan (W). Their findings revealed that an increase in the frequency of arginine in both N-terminus and C-terminus of peptides led to a significant reduction in their cytotoxicity against CHO cells compared to the sequence with a high frequency of tryptophan (W) in the N-terminal position (
48).
The ABF peptides have been found to be effective in preventing biofilm formation through the inhibition of the stringent response molecule, ppGpp (
11). In a study by Jiale et al., it was discussed that the interaction between the 1018M peptide and ppGpp was influenced by specific amino acids, with valine (V) and arginine (R) playing a crucial role in this interaction (
52). Moreover, Shang et al. demonstrated that ABF peptides containing tryptophan residues could disrupt quorum sensing and effectively inhibit biofilm formation in multidrug-resistant
Pseudomonas aeruginosa. These peptides also exhibited synergistic effects when combined with antibiotics, such as ceftazidime and piperacillin (
53).
For further investigation, a comparison was made between hiABFs and peptide sequences with only ABP effects and less than 25% ABF activity, as reported in DRAMP 2.0 and BaAMP databases (
54). Appendix 5 presents a comparison of amino acid frequencies. The analysis revealed that the frequency of lysine (K), leucine (L), arginine (R), and tryptophan (W) in hiABFs was significantly higher than ABP peptides, which lacked ABF activity or exhibited no significant activity. This analysis highlighted the significance of lysine (K), leucine (L), arginine (R), and tryptophan (W) residues in interactions with bacterial membranes and other mechanisms involved in biofilm development, such as quorum sensing and ppGpp.
An intriguing compositional analysis was conducted to compare peptide sequences that were experimentally validated to have over 50% activity against preformed biofilms (24 hours old) with sequences that influenced biofilm formation when microbial cells were exposed to them for 3 - 24 hours. The categorization of preformed and formation groups was based on the method described by Di Luca et al. (
19). Based on the results, it was observed that the formation group exhibited a higher frequency of tryptophan (W), valine (V), and arginine (R), compared to the preformed group. The AAC analysis for both groups is depicted in Appendix 6. The increased prevalence of W, V, and R amino acids in ABF peptides that function during the formation stage is probably attributable to the enhancement of interactions between peptide sequences and ppGpp or quorum-sensing molecules. The physicochemical properties, including charge, charge density, hydrophobic ratio, PI, aliphatic index, aromaticity, Boman index, and instability index of hiABFs and QSPs, were calculated; the statistical results are presented in
Figure 5.
Comparison of physicochemical properties between highly active antibiofilms (hiABFs) and quorum-sensing peptides (QSPs)
The mean positive charge of hiABFs was found to be higher than that of QSPs. A higher positive charge facilitates electrostatic interactions between peptide sequences and the negatively charged target membrane. As mentioned earlier, optimization strategies employed to enhance the antimicrobial performance of peptides against planktonic cells may not be applicable to peptides with ABF activity. Alginate is one of the major polysaccharides in the biofilm architecture of
P. aeruginosa and other pulmonary pathogens (
1). Stark et al. suggested that increasing the hydrophobicity of cationic peptides could enhance their antibacterial effects (
55). However, Benincasa et al. found that when peptide sequences were exposed to an environment containing alginate, an increase in hydrophobicity could lead to peptide aggregation and subsequent deactivation (
56).
Figure 5 illustrates a comparison of hydrophobicity between hiABFs and QSPs, revealing that hiABFs exhibited lower hydrophobicity compared to QSPs.
Moreover, when comparing hiABFs with ABPs, it was observed that the mean Eisenberg hydrophobicity for hiABFs with an experimentally confirmed high ABF activity was -0.19, while for ABPs, it was 0.11. In an experimental study focusing on IDR-1018 and 1018M peptides against methicillin-resistant
Staphylococcus aureus (MRSA), it was found that 1018M peptides inhibited biofilm formation, whereas IDR-1018 did not influence biofilm formation. Interestingly, the 1018M peptide exhibited significantly lower hydrophobicity compared to IDR-1018, with its hydrophobic ratio being 25% lower than that of IDR-1018 (
52).
Appendix 7 illustrates the comparison results of charge between ABP and ABF peptides. The analysis demonstrated that the average positive charge of ABF sequences was higher than that of antibacterial sequences. This higher positive charge in ABF peptides can be interpreted from another perspective. The biofilm matrix is known to contain extracellular DNA (eDNA) as a prominent component (
3). It has been proposed that eDNA plays a critical role in maintaining the structural integrity of biofilms (
1). In a study by Mulcahy et al., the chelator-like properties of eDNA were reported (
57). Positively charged ABF peptides exhibited a strong affinity for interacting with eDNA within the biofilm matrix. This interaction had the potential to saturate the cation-binding capacity of eDNA and potentially disrupt the eDNA-mediated resistance mechanisms of bacteria in the biofilm state (
1).
The dipeptide compositional analysis between hiABFs and QSPs highlighted several prominent dipeptide combinations, including RR, KK, RW, RI, IR, LL, LK, KL, KI, and RV, which were found to be the most abundant DPCs in hiABFs. The analysis of dipeptide compositions revealed the notable occurrence of charged and hydrophobic motifs in ABF peptides with significant ABF effects. This observation aligns with the findings reported by Bose et al., which emphasized that these dipeptide combinations reflected the amphipathic characteristics of ABFs (
58).
Figure 6 provides a comparative illustration of dipeptide compositions between hiABFs and QSPs, further elucidating the distinguishing patterns and frequencies of di-peptides in these peptide categories.
Dipeptide composition (DPC) in comparison between highly active antibiofilms (hiABFs) and quorum-sensing peptides (QSPs)
4.2. Feature Selection and Model Performance Evaluation
The performance of all 13 binary classifiers was assessed on both training and validation datasets, using 10-fold cross-validation. This process was repeated 50 times to ensure the robustness of the results. The cross-validation score, which is a reliable metric for evaluating the model performance on unseen data, was utilized for model selection. To gauge variability in performance, the standard deviation of cross-validation scores was computed and considered during the model selection process. Appendix 8 provides a summary of the overall performance of all models. The comparison of models in terms of accuracy is illustrated in Appendix 9.
Among the classifiers, the model based on SVM, random forest, and logistic regression demonstrated superior performance compared to the other classifiers. The logistic regression model achieved an accuracy of 99% on the training set and 93% on the validation set. Also, the random forest model achieved an accuracy of 99% on the training set and 94% on the validation set. Based on the findings, SVM models with different kernels (RBF, poly, and linear) showed comparable performance, with an overall accuracy of 98% on the training set and 93% on the validation set. Considering their high accuracy and consistent performance, classifiers, including logistic regression, random forest, and SVM, were selected for further optimization and analysis.
In the feature space optimization process, both filter-based and wrapper-based methods were employed. First, an MC analysis was conducted with a threshold of 0.75, resulting in the elimination of 230 highly correlated features from the initial feature space. The SelectKbest method was then applied, exploring various K values, ranging from 50 to 200. However, the best results were obtained when K was set at 100. Finally, recursive feature elimination, cross-validated (REFCV) was performed on the selected models.
Figure 7A presents the t-SNE visualization of the feature-selected data.
The perplexity parameter was set at 5.0, and the learning rate parameter was set at 200, based on empirical observations and experimentation to obtain significant visualizations. The perplexity parameter in t-SNE plays a crucial role in determining the effective number of neighbors considered for each data point during the dimensionality reduction process (
38). In the resulting t-SNE plot, clear separation between data points is not readily apparent. Despite the lack of distinct clusters, the selected classifiers were able to exploit subtle patterns and interact with feature combinations that were not visually discernible in the t-SNE plot. These classifiers successfully learned complex decision boundaries, enabling accurate predictions even in scenarios where the data points were not well-separated within the low-dimensional space. The performance of the optimized models, including the results of hyperparameter tuning and feature selection methods, was evaluated using 10-fold cross-validation on the validation set. The results of analysis, which considered different feature spaces, are presented in
Table 2.
| Model | Hyperparameters | Method of Feature Selection | Accuracy | Precision | MCC | CK | F1-Score | AUC-ROC |
|---|
| Logreg | C': 1000, 'penalty': 'l2', 'solver': 'newton-cg', 'tol': 0.01 | MO (75%) | 0.98 | 0.97 | 0.958 | 0.958 | 0.98 | 0.973 |
| REFCV | 0.99 | 0.99 | 0.986 | 0.986 | 0.99 | 0.993 |
| SelectKbest | 0.982 | 0.98 | 0.965 | 0.965 | 0.98 | 0.982 |
| RFC | 'min_samples_leaf': 2, 'n_estimators': 500, 'max_depth': 8, 'max_features': 'sqrt' | MO (75%) | 0.982 | 0.98 | 0.965 | 0.965 | 0.98 | 0.982 |
| SelectKbest | 0.97 | 0.97 | 0.947 | 0.944 | 0.97 | 0.972 |
| REFCV | 0.97 | 0.97 | 0.9375 | 0.9375 | 0.97 | 0.9688 |
| SVM_rbf | 'C': 100, 'gamma': 0.001 | MO (75%) | 0.985 | 0.99 | 0.971 | 0.979 | 0.99 | 0.98 |
| SelectKbest | 0.97 | 0.975 | 0.944 | 0.942 | 0.97 | 0.973 |
| SVM_poly | 'C': 0.1, 'degree': 2, 'gamma': 1 | MO (75%) | 0.98 | 0.96 | 0.958 | 0.958 | 0.98 | 0.978 |
| SelectKbest | 0.98 | 0.98 | 0.953 | 0.958 | 0.98 | 0.948 |
| SVM_lnr | 'C': 0.1, 'gamma': 1 | MO (75%) | 0.98 | 0.99 | 0.972 | 0.972 | 0.99 | 0.986 |
| REFCV | 0.99 | 0.99 | 0.976 | 0.979 | 0.99 | 0.99 |
| SelectKbest | 0.96 | 0.96 | 0.916 | 0.916 | 0.96 | 0.957 |
Abbreviations: Logreg, logistic regression; RFC, random forest classifier; SVM, support vector machine; MCC, Matthew's correlation coefficient; CK, Cohen’s kappa statistic; AUC-ROC, area under the receiver operating characteristic curve; REFCV, recursive feature elimination, cross-validated.
In
Figure 7B, the ROC curves for models with superior performance are displayed. These curves were plotted using the Orange 3.33.0 Python package (
59).
The t-distributed stochastic neighbor embedding (t-SNE) visualization of feature-selected data (A); the ROC curve illustrates the ability of the selected classifier to distinguish between highly active antibiofilms (hiABFs) and quorum-sensing peptides (QSPs).
Diverse metrics were used for the analysis and comparison of model performance. The numbers of positive and negative libraries were 183 and 198, respectively. Consequently, the dataset sizes were comparable and almost balanced, enabling the use of accuracy as a criterion for assessing the model performance. To mitigate the potential for overly optimistic reporting of model performance, the MCC value was also taken into consideration. Equation 12 demonstrates that MCC yields a high score only when the binary classifiers perform well in all categories of the confusion matrix, including TP, FN, TN, and FP, proportionally adjusted to the size of positive and negative libraries (
41).
Table 2 presents the results of analysis based on the MCC value, which demonstrate the strong performance of classifiers in predicting the high ABF activity of peptide sequences.
4.4. Conclusions
In recent decades, the characteristics of infections have undergone fundamental changes, primarily due to antibiotic resistance caused by bacterial biofilms. Consequently, a significant emphasis has been placed on developing antimicrobial agents that can address the challenges posed by antibiotic resistance. Peptides have emerged as a promising class of antimicrobial biomolecules, with ABF peptides showing remarkable potential in eradicating preformed biofilms or inhibiting biofilm formation. Meanwhile, experimental methods for designing and synthesizing ABF peptides can be cumbersome. Therefore, development of computational methods to streamline this process has become indispensable. The application of machine learning and artificial intelligence in drug discovery and development has gained significant attention due to its advantages. Consequently, there has been a rapid increase in the number of studies utilizing these techniques for peptide prediction and design. In this study, the advantages of multiple machine learning algorithms were utilized to create a computational platform for predicting the high ABF effect of peptide sequences. The focus was on ABF peptides with significant activity due to a lack of research incorporating significant ABF activity in their datasets and model development. The dataset gathering process involved the inclusion of peptide sequences with ABF activity equal to or higher than 50%. As for the negative datasets, QSPs were utilized. Duplicated sequences, sequences containing non-standard amino acid residues, and sequences with any modifications or indefinite residues were excluded from the datasets. The feature space was created by calculating features based on physicochemical properties, amino acid composition, order, and their distribution. Filter- and wrapper-based feature selection methods were employed to construct the feature space. A range of binary classifiers with 10-fold cross-validation was used to identify models with superior performance, which were subsequently optimized by adjusting their hyperparameters using GridSearchCV. Among the selected models, those based on logistic regression, SVM, and random forest demonstrated better performance on both training and validation datasets in terms of accuracy, precision, and MCC. The model performance and the created feature space were evaluated on an independent test set to predict highly active ABF properties. The model achieved 99% accuracy, 99% precision, and an MCC of 0.979. Overall, an in-depth analysis of the structure, composition, amino acid preferences, and relationship with the mechanism of action in hiABFs can greatly facilitate the design of peptide-based therapeutics. While computational methods play a crucial role in streamlining the drug design and development process, it is important to acknowledge that there is a long way ahead before computer algorithms can lead to the development of FDA-approved drugs.
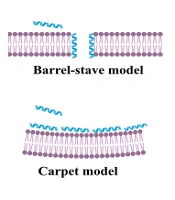




; two main mechanisms of action of antibiofilm (ABF) agents include membrane-lytic peptides (B); and interference with ppGpp signaling process (C)")

 between highly active antibiofilms (hiABFs) and quorum-sensing peptides (QSPs); B, the figure demonstrates the distribution of aliphatic, aromatic, positively charged, negatively charged, and uncharged amino acids in hiABFs and QSPs.")
; and B, quorum-sensing peptides (QSPs). The size of residues is proportional to their propensity.")
 and quorum-sensing peptides (QSPs)")
 in comparison between highly active antibiofilms (hiABFs) and quorum-sensing peptides (QSPs)")
 visualization of feature-selected data (A); the ROC curve illustrates the ability of the selected classifier to distinguish between highly active antibiofilms (hiABFs) and quorum-sensing peptides (QSPs).")