1. Background
In recent years, Candida albicans has emerged as an important fungal pathogen and a common opportunistic agent in immunocompromised patients. Candida albicans has led to the emergence of both superficial and deep infections in the past 3 decades (1-3). Considering the rising prevalence of immunosuppression, long-term hospitalization, and invasive medical treatments, Candida species have become a major group of opportunistic pathogens causing infections in humans. Prevention and treatment of candidiasis are of importance considering the prevalence of nosocomial Candida infections (4). Recurrent or persistent infections caused by Candida species are common, especially among patients with vaginal and oropharyngeal candidiasis (OPC); nonetheless, such infections have also been reported in patients with urinary tract infections (5, 6).
Through genotyping of C. albicans, we can reach the following goals: 1) determining the relationship between commensalism and infection; 2) identifying nosocomial candidiasis to determine strains associated with the outbreak of infection; 3) distinguishing epidemic from sporadic or endemic strains; 4) identifying the etiology of infection, strain transmission, and acquisition routes; 5) examining the genetic diversity of isolates from a carrier; 6) determining recurrent infections to find virulent strains; 7) evaluating the emergence of drug-resistant strains; and 8) assessing the population diversity, structure, and dynamics (7, 8). Typing methods are recognized as helpful tools in differentiating strains causing recurrent infections (7). Despite the availability of different typing methods for C. albicans (eg, PCR-RFLP, AFLP, and MLST), microsatellite length polymorphism (MLP) is known as a highly efficient method of fragment length analysis of microsatellites (9, 10), with high reproducibility and discriminatory power (11).
Various epidemiological studies have confirmed the reproducibility and efficacy of MLP analysis. As a PCR-based method, MLP typing uses high repeat variability in microsatellite sequences (described as tandem repeats of 2 - 6 nucleotides). Microsatellite markers include a definite pair of primers flanking a particular microsatellite site inside the genome. For amplifying specific loci, fluorescence-labeled primers are used, and the allele length is measured via gel electrophoresis of PCR products with an automatic sequencer. In general, MLP is a highly discriminative method for C. albicans typing. Nevertheless, the microsatellite marker affects its resolving power. In the C. albicans genome, different polymorphic microsatellite loci have been described (eg, CDC3, EF3, HIS3, ERK1; 2NF1, CCN2, EFG1, CPH2, CAI; and CAIII-CAVII). A combination of markers on different chromosomes from the same typing system facilitates a more precise classification of C. albicans with MLP (10, 12, 13).
2. Objectives
In this study, we aimed at examining the genetic diversity and relatedness among isolates of C. albicans, collected from HIV positive patients with oral candidiasis, using microsatellite fragment length polymorphism.
3. Methods
A total of 30 C. albicans isolates were collected from patients with HIV and OPC in Tehran, Iran. Moreover, C. albicans was isolated from 8 out of 30 patients with recurrent OPC in 2 different episodes. To identify the origin of cutaneous and nail infections in six patients, C. albicans isolates, as well as isolates related to OPC were analyzed. C. albicans isolates were collected from CHROMagar Candida and Sabouraud dextrose agar media after incubation for 72 hours at 35°C. Phenotypic identification was confirmed using different tests including chlamydospore production, colony morphology on CHROM agar, and carbohydrate assimilation using rapid yeast identification system (Remel Inc., USA) (14, 15).
For DNA extraction, a physicochemical method was applied. A portion of the yeast colony was washed with phosphorous-buffered saline containing 50 mM EDTA and 0.5% SDS. Afterwards, the freeze-thawing method with glass beads was applied to disrupt the yeasts. After centrifugation for 2 minutes at 12,000 g, lysis buffer (500 mL) was added to the precipitate, followed by incubation for 10 minutes. Potassium acetate buffer (150 µL, pH = 4.8; 28.5 mL of distilled water; 60 mL of 5 M potassium acetate; 11.5 mL of glacial acetic acid) was then added to the tube and vortexed for a short period.
Centrifugation (2 minutes at 12,000 g) was used to remove the precipitated proteins and cell debris. The supernatant was transferred to a microtube and centrifuged for 2 minutes at 12,000 g. After decanting the supernatant to a new microtube (1.5 mL), isopropyl alcohol was added with an equal volume. The tube was briefly mixed via inversion and centrifuged for 2 minutes at 12,000 g. After removing the supernatant, the DNA pellet was washed 3 times in 70% ethanol (300 µL). The supernatant was discarded after centrifugation for 1 minute at 12,000 g. Then, DNA was dried and dissolved in distilled water (50 µL).
To determine DNA concentration and its purification, optical density (OD) was read and run on agarose gel. To amplify the 3 loci, multiplex PCR was performed in a reaction volume of 50 µL, containing 5 mM of MgCl2, 1X PCR buffer, 0.2 mM of each dNTP, 1.25 U of Taq gold polymerase (Applied Biosystems), 5 pmol of EF3 (Chromosome 5) primers, and 2 pmol of CDC3 (Chromosome 1), and HIS3 (chromosome 2) primers. The microsatellite markers were amplified with the primers (Table 1), and different dyes were used to label a primer from each set. The EF3 antisense primer was stained using fluorescein amidite (FAM), CDC3 sense primer was labeled with hexachlorofluorescein (HEX), and HIS3 sense primer was labeled with NED (16). The PCR included initial denaturation at 95°C for 10 minutes, followed by 30 cycles at 95°C for 30 seconds, at 55°C for 30 seconds, and at 72°C for 60 seconds, followed by a final extension at 72°C for 30 minutes. After amplification, 2 µL of the PCR product was added to formamide (20 µL) and 6-TAMRA Genescan 500 size standard (0.5 µL, Applied Biosystems). The samples were run on an ABI XL 370 genetic analyzer after denaturation at 95°C for 120 seconds and were placed in an ice bath. Moreover, the GeneMapper Version 5 (Applied Biosystems) was used to determine the band size (allelic number) (13, 16, 17).
Table 1.Details of Microsatellite Markers Applied in the Molecular Analysis of C. albicans
| Locus, Chromosome | Gene Product | Primer | Sequence 5′ - 3′ | Dye | Repeat Type | Size Range |
|---|---|---|---|---|---|---|
| EF3, chr 5 | Elongation factor 3 | EF3F | TTTCCTCTTCCTTTCATATAGAA | FAM | TTTC-TTC | 120 - 141 |
| EF3R | GGATTCACTAGCAGCAGACA | |||||
| CDC3, chr 1 | Cell division cycle protein | CDC3F | CAGATGATTTTTTGTATGAGAAGAA | HEX | AGTA | 112 - 140 |
| CDC3R | CAGTCACAAGATTAAAATGTTCAAG | |||||
| HIS3, chr 2 | Imidazole glycerol | HIS3F | TGGCAAAAATGATATTCCAA | NED | TTG | 224 - 245 |
| HIS3R | TACACTATGCCCCAAACACA |
4. Results
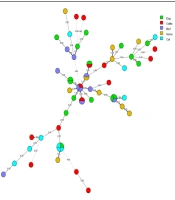
There were I or 2 bands based on the multiplex PCR analysis of each microsatellite marker and isolate. As C. albicans is a diploid pathogen, every marker had a single locus, and every band was attributed to an allele. Accordingly, a profile of 6 alleles characterized every isolate. Overall, 27 different profiles were obtained via PCR MLP typing of 30 isolates (discriminatory power: 0.993) (Figure 1). There were 10 alleles and 18 combinations, 10 alleles and 14 combinations, and 7 alleles and 18 combinations for EF3, HIS3, and EF3 genes, respectively. Heterogeneity was observed in allele frequency. In a particular locus, the most probabilistic number of repeats indicated an association with the increase or decrease in the number of repeats. Nevertheless, the alleles were not normally distributed, and some were overrepresented. Furthermore, most isolates were heterozygous inside at least 1 locus, whereas only 2 isolates were homozygous in all the 3 loci (Table 2). Although the haploidy of these isolates was not definite, they were described as homozygous.
Table 2.Demographic Data and Genotypes of 30 Clinical C. albicans Isolates Collected from HIV Positive Patients
| Isolates Number | Isolates Origin | Fragments Length (Bp) Identified By Gene Scan | Final Genotype | Fluconazole resistance | Gender | CD4 Count | HAART | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EF3 marker | CDC3 marker | HIS3 marker | ||||||||||
| Allele1 | Allele2 | Allele1 | Allele2 | Allele1 | Allele2 | |||||||
| N1 | OPC | 129 | 136 | 116 | 116 | 150 | 162 | GT9 | DD | M | < 200 | No |
| N10 | OPC | 129 | 138 | 116 | 128 | 154 | 154 | GT1 | S | F | < 200 | No |
| N12 | OPC | 123 | 123 | 120 | 120 | 154 | 154 | GT2 | S | F | < 200 | No |
| N13 | OPC | 129 | 138 | 116 | 116 | 154 | 154 | GT3 | S | F | < 200 | No |
| N14 | OPC | 129 | 137 | 124 | 128 | 150 | 167 | GT4 | DD | F | < 200 | No |
| N15 | OPC | 129 | 138 | 116 | 128 | 154 | 154 | GT5 | S | M | < 200 | No |
| N16 | OPC | 129 | 129 | 116 | 116 | 162 | 162 | GT6 | S | M | < 200 | No |
| N17 | OPC | 129 | 141 | 116 | 128 | 154 | 154 | GT7 | R | M | < 200 | No |
| N18 | OPC | 129 | 138 | 128 | 128 | 154 | 154 | GT8 | S | M | < 200 | No |
| N22 | OPC | 138 | 138 | 116 | 140 | 154 | 154 | GT10 | S | M | < 200 | No |
| N24 | OPC | 123 | 129 | 112 | 116 | 150 | 162 | GT11 | R | M | < 200 | Yes |
| N27 | OPC | 123 | 129 | 116 | 124 | 180 | 180 | GT12 | R | M | < 200 | Yes |
| N28 | OPC | 123 | 129 | 112 | 116 | 150 | 162 | GT11 | S | M | 200 - 400 | No |
| N29 | OPC | 129 | 138 | 128 | 128 | 154 | 154 | GT13 | R | M | 200 - 400 | No |
| N30 | OPC | 120 | 123 | 120 | 120 | 154 | 154 | GT14 | S | F | 200 - 400 | No |
| N31 | OPC | 129 | 137 | 124 | 128 | 150 | 166 | GT15 | S | M | 200 - 400 | Yes |
| N33 | OPC | 124 | 136 | 120 | 124 | 154 | 192 | GT16 | R | M | 200 - 400 | Yes |
| N35 | OPC | 123 | 137 | 120 | 124 | 154 | 166 | GT17 | S | F | 200 - 400 | Yes |
| N42 | OPC | 120 | 128 | 116 | 124 | 162 | 162 | GT18 | S | F | 200 - 400 | Yes |
| N44 | OPC | 124 | 136 | 120 | 120 | 154 | 172 | GT19 | S | M | 200 - 400 | Yes |
| N46 | OPC | 129 | 136 | 116 | 116 | 149 | 162 | GT20 | S | M | 200 - 400 | Yes |
| N47 | OPC | 124 | 138 | 112 | 116 | 149 | 149 | GT21 | S | M | 200 - 400 | Yes |
| N51 | OPC | 123 | 138 | 116 | 116 | 179 | 179 | GT22 | S | F | > 400 | Yes |
| N54 | OPC | 123 | 137 | 120 | 124 | 154 | 166 | GT23 | R | M | > 400 | Yes |
| N55 | OPC | 129 | 136 | 116 | 116 | 150 | 162 | GT9 | R | M | > 400 | Yes |
| N56 | OPC | 120 | 120 | 124 | 124 | 162 | 162 | GT24 | R | M | > 400 | Yes |
| N57 | OPC | 129 | 140 | 116 | 116 | 150 | 162 | GT25 | S | M | > 400 | Yes |
| N58 | OPC | 120 | 125 | 124 | 124 | 147 | 166 | GT26 | DD | M | > 400 | No |
| N59 | OPC | 129 | 135 | 116 | 128 | 154 | 154 | GT27 | S | M | > 400 | Yes |
| N60 | OPC | 129 | 141 | 116 | 116 | 154 | 154 | GT7 | R | M | > 400 | Yes |
Abbreviations: DD, dose dependent susceptible; OPC, Oropharyngeal candidiasis, R, resistant S, susceptible.
To identify the origin of cutaneous and nail infections among 6 patients, C. albicans isolates from the culture, as well as isolates related to oral cavity infections were genotyped. The genotypes were identified in 4 of these patients. The genotype of C. albicans from the nails differed from the genotype of oral cavity in 2 patients and was considered as an exogenous source of superficial infection. To evaluate microevolution and change the isolates in recurrent OPC, C. albicans was isolated from the culture of 8 patients and genotyped in 2 different episodes of disease in different intervals. The genotypes were identical in 6 patients. In addition, changes in only 1 or 2 alleles, which represent microevolution (evolutionary changes within a species or small group of organisms, especially over a short period), were observed in some isolates (Table 3).
Table 3.Comparison of 8 Pairs of Isolates from Recurrent Oral Collected at Different Time from Same Patients Underlying Alleles Related to Change in C. albicans Genotypes
| Patients/Episode | Length, bp Determined by PCR Analysis | |||||
|---|---|---|---|---|---|---|
| EF3 | CDC3 | HIS3 | ||||
| Allele1 | Allele2 | Allele1 | Allele2 | Allele1 | Allele2 | |
| P1 | ||||||
| Episode1 | 129 | 138 | 116 | 128 | 154 | 154 |
| Episode2 | 129 | 138 | 116 | 128 | 154 | 154 |
| P2 | ||||||
| Episode1 | 129 | 138 | 116 | 116 | 154 | 154 |
| Episode2 | 129 | 138 | 116 | 128 | 154 | 154 |
| P3 | ||||||
| Episode1 | 120 | 123 | 120 | 120 | 154 | 154 |
| Episode2 | 120 | 123 | 120 | 120 | 154 | 154 |
| P4 | ||||||
| Episode1 | 120 | 128 | 116 | 124 | 162 | 162 |
| Episode2 | 120 | 120 | 124 | 124 | 162 | 162 |
| P6 | ||||||
| Episode1 | 123 | 137 | 120 | 124 | 154 | 166 |
| Episode2 | 123 | 137 | 120 | 124 | 154 | 166 |
| P7 | ||||||
| Episode1 | 124 | 136 | 120 | 124 | 154 | 192 |
| Episode2 | 124 | 136 | 120 | 124 | 154 | 192 |
| P8 | ||||||
| Episode1 | 129 | 141 | 116 | 116 | 154 | 154 |
| Episode2 | 129 | 141 | 116 | 116 | 154 | 154 |

Figure 1.
Genetic Diversity of 30 Dependent C. albicans Isolates Collected from HIV Positive Patients Via MLP Typing
5. Discussion
Candida albicans is capable of causing severe infections, particularly in immunocompromised patients. Consequently, researchers have attempted to develop accurate methods to discriminate between isolates. Understanding the relationship between the strains involved in Candida infections is strongly associated with the development of proper treatment methods and adds to the available epidemiological knowledge. Epidemiological studies have applied different typing methods for characterizing C. albicans isolates including electrophoretic karyotyping, southern blot hybridization using discriminating probes, RFLP, and random polymorphic DNA amplification. Microsatellites are common markers for differentiating between microorganisms, given their potential application and high diversity in automated tests besides the simplicity of PCR amplification and interpretation of the results (18, 19). Candida albicans isolates contain many loci, and accordingly, several markers have been developed for them (16, 19). However, it seems that most of microsatellites in coding regions have low levels of heterozygosity, although the use of multiplex PCR and the evaluation of more loci increase the discriminatory power (20, 21).
The analysis of 30 independent isolates recovered from HIV-positive patients with 3 loci (CDC3, EF3, and HIS3) identified 27 different genotypes (discriminatory power of 0.993). The results of multiplex PCR regarding genetic relatedness discriminated 27 out of 30 (90%) strains, which otherwise would have been considered similar. Microevolution was reported due to minor changes in the strain genotypes including changes in only 1 allele, which could be described in a single mutational step. Using this method to assess recurrent infection isolates shows similar scenarios as previously described. Among 8 recurrent cases, 6 were related to a similar strain, and 2were attributed to a similar strain undergoing microevolution (One allele was changed.). Furthermore, MLP could detect microevolutionary changes and was useful in identifying microevolution after environmental stress. Moreover, it can improve therapeutic procedures in patients due to the recurrence of refractory fungal infections. The primers, analysis techniques, and allele nomenclature for microsatellite typing systems should be standardized. Therefore, for interlaboratory comparisons, these important issues need to be considered. Moreover, the development of a global public database similar to MLST is essential for the collection of microsatellite allele data. The present study can help develop a genetic database of fungal pathogens in Iran. A relatively high divergence was found in the structure of C. albicans among HIV-positive patients. The isolates were classified into 27 genotypes, showing major redundancy in isolates from clinical samples.
In this study, various strains with similar MLP genotypes were reported (30 isolates from 27 genotypes) (Figure 1). We found isolates, which were only different in 1allele and might have evolved from a common ancestor. No association was found between fluconazole resistance and genotypes. However, to determine the molecular epidemiology of C. albicans in Iran, further research is required on C. albicans strains in other regions of the country. Based on the present findings, potentially pathogenic C. albicans have the opportunity to overgrow in the context of HIV infection. In some previous studies, mixed types were detected in some patients. In the host, the strains undergo microvariation, which often involves alterations in zygosity of diploid allele pairs, but show inadequate geographical relatedness (2, 22, 23).
6. Conclusions
Considering the high variability of genotypes in the population of C. albicans, microsatellite fragment length polymorphism analysis with multiplex PCR facilitates high-speed genotyping for molecular epidemiological evaluation.