


1. Background
2. Objectives

Glycyrrhetinic acid induces autophagy by inhibiting the TGF-β1/SMAD signaling pathway, thereby promoting anti-tumor macrophage polarization and ultimately suppressing the malignant progression of hepatocellular carcinoma.
3. Methods
3.1. Cell Culture and Induction
3.2. Cell Treatment
3.3. Animals and Treatment
3.4. Cell Co-culture
3.5. Cell Counting Kit-8 Assay
3.6. mRFP-GFP-LC3B Viral Transfection
3.7. EdU Staining
3.8. Scratch Assay
3.9. Transwell Assay
3.10. Immunofluorescence
3.11. Immunohistochemistry
3.12. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling Staining
3.13. Hematoxylin and Eosin Staining
3.14. Enzyme-Linked Immunosorbent Assay
3.15. Flow Cytometry
3.16. Western Blot
3.17. Statistical Analysis
4. Results
4.1. Glycyrrhetinic Acid Inhibits Proliferation, Migration, Invasion, and Promotes Apoptosis of HCC Cells
 inhibits the proliferation, migration, and invasion of hepatocellular carcinoma (HCC) cells while promoting apoptosis. A, chemical structure of GA; B, effect of different concentrations of GA (0/12.5/25/50/100/150/200 μM) on the viability of human normal hepatocyte THLE-2 cells after 24 h treatment, as determined by cell counting kit-8 (CCK-8) assay; C-E, effect of different concentrations of GA (0/12.5/25/50/100/150 μM) on the viability of Li-7, HuH-7, and HCC-LM3 after 24 h and 48 h treatment, respectively, as determined by CCK-8 assay; F-H, effect of GA treatment for 24 h on the proliferation of HCC cells, analyzed by EdU staining (scale bar: 50 μm); I-K, effect of GA treatment for 24 h on the migration of HCC cells, determined by scratch assay (scale bar: 200 μm); L-N, effect of GA treatment for 24 h on the invasion of HCC cells, analyzed by Transwell invasion assay (scale bar: 100 μm); O-Q, effect of GA treatment for 24 h on the apoptosis of HCC cells, detected by flow cytometry (ns: P > 0.05, * P < 0.05, ** P < 0.01, *** P < 0.001 vs control).")
Glycyrrhetinic acid (GA) inhibits the proliferation, migration, and invasion of hepatocellular carcinoma (HCC) cells while promoting apoptosis. A, chemical structure of GA; B, effect of different concentrations of GA (0/12.5/25/50/100/150/200 μM) on the viability of human normal hepatocyte THLE-2 cells after 24 h treatment, as determined by cell counting kit-8 (CCK-8) assay; C-E, effect of different concentrations of GA (0/12.5/25/50/100/150 μM) on the viability of Li-7, HuH-7, and HCC-LM3 after 24 h and 48 h treatment, respectively, as determined by CCK-8 assay; F-H, effect of GA treatment for 24 h on the proliferation of HCC cells, analyzed by EdU staining (scale bar: 50 μm); I-K, effect of GA treatment for 24 h on the migration of HCC cells, determined by scratch assay (scale bar: 200 μm); L-N, effect of GA treatment for 24 h on the invasion of HCC cells, analyzed by Transwell invasion assay (scale bar: 100 μm); O-Q, effect of GA treatment for 24 h on the apoptosis of HCC cells, detected by flow cytometry (ns: P > 0.05, * P < 0.05, ** P < 0.01, *** P < 0.001 vs control).
4.2. Glycyrrhetinic Acid Inhibits pro-tumorigenic Polarization of Tumor-Associated Macrophages, Thereby Suppressing Hepatocellular Carcinoma Cell Viability and Promoting Apoptosis
 after GA treatment, detected by enzyme-linked immunosorbent assay; S-T, Proliferation of HCC cells detected by EdU staining (scale bar: 50 μm); U-V, invasion of HCC cells detected by Transwell assay (scale bar: 100 μm); W-X, migration of HCC cells detected by scratch assay (scale bar: 200 μm); Y-Z, apoptosis of HCC cells detected by flow cytometry (ns, P > 0.05, * P < 0.05, ** P < 0.01, *** P < 0.001 vs control).")
Glycyrrhetinic acid inhibits M2 polarization of TAMs, thereby suppressing HCC cell viability and promoting apoptosis. A, identification of successful differentiation of THP-1 cells into M0 macrophages by detecting CD11b and CD14 expression via flow cytometry; B, identification of successful differentiation of THP-1 cells into M1 macrophages by detecting CD11b and CD86 expression via flow cytometry; C, identification of successful differentiation of THP-1 cells into M2 macrophages by detecting CD11b and CD206 expression via flow cytometry; D-G, western blot analysis of the effect of GA on M1 marker protein expression in M1 macrophages; H-K, western blot analysis of the effect of GA on M2 marker protein expression in M2 macrophages; L-O, low cytometry validation of GA's effect on M1/M2 macrophage polarization status. CD86 positive indicates M1 macrophages. CD206 positive indicates M2 macrophages; P, schematic diagram of the co-culture system for HCC cells and M2 macrophages; Q-R, secretion levels of M2 macrophage-related cytokines (IL-10, TGF-β1) after GA treatment, detected by enzyme-linked immunosorbent assay; S-T, Proliferation of HCC cells detected by EdU staining (scale bar: 50 μm); U-V, invasion of HCC cells detected by Transwell assay (scale bar: 100 μm); W-X, migration of HCC cells detected by scratch assay (scale bar: 200 μm); Y-Z, apoptosis of HCC cells detected by flow cytometry (ns, P > 0.05, * P < 0.05, ** P < 0.01, *** P < 0.001 vs control).
4.3. Glycyrrhetinic Acid Induces Autophagy in Hepatocellular Carcinoma Cells, Thereby Interfering with Pro-tumorigenic Polarization of Tumor-Associated Macrophages
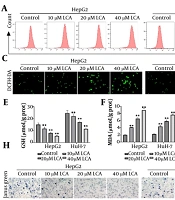
. The effect of GA on autophagic flux was observed by analyzing fluorescent puncta in HepG2 and SMMC-7721 cells transfected with mRFP-GFP-LC3B virus. Yellow puncta (colocalization of mRFP and GFP) represent autophagosomes not fused with lysosomes; orange-red puncta (mRFP signal only) represent autolysosomes fused with lysosomes (scale bar: 50 μm); G-H, viability of HCC cells detected by cell counting kit-8 assay; I-N, co-culture of HCC cells (control, 150 μM GA, 150 μM GA + 10 μM CQ) with M2 macrophages in a Transwell system to detect changes in the M2 macrophage phenotype; I-J, secretion levels of M2 macrophage-related cytokines IL-10 and TGF-β1 in culture supernatant detected by enzyme-linked immunosorbent assay; K-N, protein expression levels of M2 macrophage markers CD206, IL-10, and Arg1 detected by Western blot (* P < 0.05, ** P < 0.01, *** P < 0.001 vs control; # P < 0.05, ## P < 0.01 vs 150 μM GA).")
Glycyrrhetinic acid induces autophagy in HCC cells, thereby interfering with M2 polarization of TAMs. A-C, to investigate the effect of GA on autophagy, this study detected its impact on the expression of key autophagy marker proteins in HCC cells via Western blot; D-F, experimental groups: Control, 150 μM GA, and co-treatment with 150 μM GA and 10 μM chloroquine (CQ). The effect of GA on autophagic flux was observed by analyzing fluorescent puncta in HepG2 and SMMC-7721 cells transfected with mRFP-GFP-LC3B virus. Yellow puncta (colocalization of mRFP and GFP) represent autophagosomes not fused with lysosomes; orange-red puncta (mRFP signal only) represent autolysosomes fused with lysosomes (scale bar: 50 μm); G-H, viability of HCC cells detected by cell counting kit-8 assay; I-N, co-culture of HCC cells (control, 150 μM GA, 150 μM GA + 10 μM CQ) with M2 macrophages in a Transwell system to detect changes in the M2 macrophage phenotype; I-J, secretion levels of M2 macrophage-related cytokines IL-10 and TGF-β1 in culture supernatant detected by enzyme-linked immunosorbent assay; K-N, protein expression levels of M2 macrophage markers CD206, IL-10, and Arg1 detected by Western blot (* P < 0.05, ** P < 0.01, *** P < 0.001 vs control; # P < 0.05, ## P < 0.01 vs 150 μM GA).
4.4. GA Inhibits the TGF-β1/SMAD Pathway
 (* P < 0.05, ** P < 0.01, *** P < 0.001 vs control).")
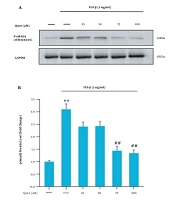
Glycyrrhetinic acid inhibits the TGF-β1/SMAD pathway. A-D, western blot analysis of the effect of GA treatment on the expression levels of key proteins in the TGF-β1/SMAD pathway in HCC cells; E-F, immunofluorescence staining validation of GA's effect on TGF-β1 protein expression in HCC cells (scale bar: 50 μm) (* P < 0.05, ** P < 0.01, *** P < 0.001 vs control).
4.5. Glycyrrhetinic Acid Induces Autophagy by Inhibiting the Activation of the TGF-β1/SMAD Pathway
 induces autophagy by inhibiting the activation of the TGF-β1/SMAD pathway. A-D, experimental groups: Control, 150 μM GA, 150 μM GA + 10 μM SRI-011381. Western blot analysis of the expression and phosphorylation levels of key proteins in the TGF-β1/SMAD signaling pathway in HCC cells; E-G, effect of SRI-011381 on GA-induced autophagic flux analyzed by fluorescent puncta using mRFP-GFP-LC3B viral transfection. Yellow puncta represent autophagosomes, and orange-red puncta represent autolysosomes (scale bar: 50 μm); H-J, the expression levels of autophagy marker proteins in cells from each group (*** P < 0.001 vs control; # P < 0.05 vs 150 μM GA).")
Glycyrrhetinic acid (GA) induces autophagy by inhibiting the activation of the TGF-β1/SMAD pathway. A-D, experimental groups: Control, 150 μM GA, 150 μM GA + 10 μM SRI-011381. Western blot analysis of the expression and phosphorylation levels of key proteins in the TGF-β1/SMAD signaling pathway in HCC cells; E-G, effect of SRI-011381 on GA-induced autophagic flux analyzed by fluorescent puncta using mRFP-GFP-LC3B viral transfection. Yellow puncta represent autophagosomes, and orange-red puncta represent autolysosomes (scale bar: 50 μm); H-J, the expression levels of autophagy marker proteins in cells from each group (*** P < 0.001 vs control; # P < 0.05 vs 150 μM GA).
4.6. Glycyrrhetinic Acid Regulates the TGF-β1/SMAD Pathway to Induce Autophagy in Cancer Cells In Vivo and Inhibit Tumor Growth
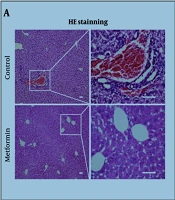
 regulates the TGF-β1/SMAD pathway to induce autophagy in cancer cells in vivo and inhibit tumor growth. A-H, experimental groups: Control group, GA (25 mg/kg) group, GA (50 mg/kg) group, GA (50 mg/kg) + SRI-011381 (30 mg/kg) group (n = 9). Western blot analysis of the expression levels of key proteins in the TGF-β1/SMAD axis and autophagy marker proteins in mouse tumor tissues; I-K, evaluation of GA's inhibitory effect on tumor growth in mice; I, tumor volume change curve during treatment (days 0, 7, 14, 21, 28); J, representative photographs of tumors excised on day 28; K, tumor weight measured on day 28; L-M, expression levels of the proliferation marker Ki67 in mouse tumor tissues detected by immunohistochemistry staining; N-O, apoptosis levels of tumor cells detected by terminal deoxynucleotidyl transferase dUTP nick end labeling staining; P, pathological changes in mouse liver and kidney tissues observed by hematoxylin and eosin staining (scale bar: 50 μm) (* P < 0.05, ** P < 0.01, *** P < 0.001 vs control; # P < 0.05, ## P < 0.01 vs 50 mg/kg GA).")
Glycyrrhetinic acid (GA) regulates the TGF-β1/SMAD pathway to induce autophagy in cancer cells in vivo and inhibit tumor growth. A-H, experimental groups: Control group, GA (25 mg/kg) group, GA (50 mg/kg) group, GA (50 mg/kg) + SRI-011381 (30 mg/kg) group (n = 9). Western blot analysis of the expression levels of key proteins in the TGF-β1/SMAD axis and autophagy marker proteins in mouse tumor tissues; I-K, evaluation of GA's inhibitory effect on tumor growth in mice; I, tumor volume change curve during treatment (days 0, 7, 14, 21, 28); J, representative photographs of tumors excised on day 28; K, tumor weight measured on day 28; L-M, expression levels of the proliferation marker Ki67 in mouse tumor tissues detected by immunohistochemistry staining; N-O, apoptosis levels of tumor cells detected by terminal deoxynucleotidyl transferase dUTP nick end labeling staining; P, pathological changes in mouse liver and kidney tissues observed by hematoxylin and eosin staining (scale bar: 50 μm) (* P < 0.05, ** P < 0.01, *** P < 0.001 vs control; # P < 0.05, ## P < 0.01 vs 50 mg/kg GA).
4.7. Glycyrrhetinic Acid Inhibits M2 Macrophage Polarization in vivo by Regulating the TGF-β1/SMAD Pathway
 inhibits M2 macrophage polarization in vivo by regulating the TGF-β1/SMAD pathway. A-D, flow cytometry analysis of macrophage polarization in mouse tumor tissues; A-B, proportion of F4/80⁺ total macrophages among tumor-infiltrating immune cells; C-D, proportion of CD206⁺ M2 macrophages among F4/80⁺ cells; E-F, in vitro co-culture system to validate the direct effect of GA on macrophage polarization. Mouse BMDMs were co-cultured with mouse HCC cells Hepa1-6 in a Transwell system. Enzyme-linked immunosorbent assay detection of secretion levels of M2 macrophage-related cytokines IL-10 and TGF-β1 in the co-culture supernatant; G-J, western blot analysis of the expression levels of M2 marker proteins (CD206, IL-10, Arg1) in BMDMs; K-L, proportion of CD206⁺ cells in BMDMs within the co-culture system detected by flow cytometry (** P < 0.01, *** P < 0.001 vs control; # P < 0.05, ## P < 0.01 vs 50 mg/kg GA).")
Glycyrrhetinic acid (GA) inhibits M2 macrophage polarization in vivo by regulating the TGF-β1/SMAD pathway. A-D, flow cytometry analysis of macrophage polarization in mouse tumor tissues; A-B, proportion of F4/80⁺ total macrophages among tumor-infiltrating immune cells; C-D, proportion of CD206⁺ M2 macrophages among F4/80⁺ cells; E-F, in vitro co-culture system to validate the direct effect of GA on macrophage polarization. Mouse BMDMs were co-cultured with mouse HCC cells Hepa1-6 in a Transwell system. Enzyme-linked immunosorbent assay detection of secretion levels of M2 macrophage-related cytokines IL-10 and TGF-β1 in the co-culture supernatant; G-J, western blot analysis of the expression levels of M2 marker proteins (CD206, IL-10, Arg1) in BMDMs; K-L, proportion of CD206⁺ cells in BMDMs within the co-culture system detected by flow cytometry (** P < 0.01, *** P < 0.001 vs control; # P < 0.05, ## P < 0.01 vs 50 mg/kg GA).