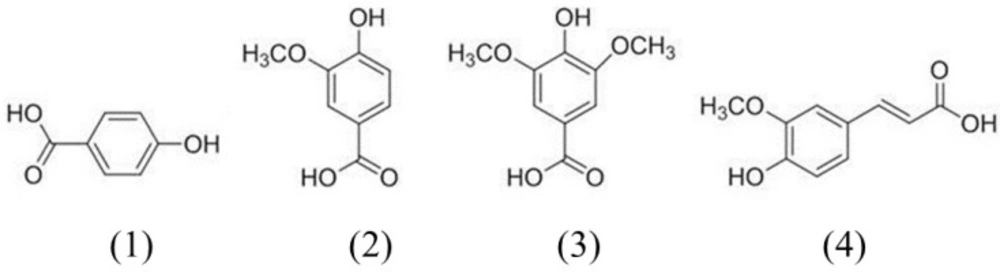
Chemicals
4-Hydroxybenzoic acid (purity, ≥99.0%), syringic acid (≥95.0%), vanillic acid (≥97.0%), cisplatin (≥99.0%), captopril (≥98.0%), and crystal violet solution were purchased from Sigma-Aldrich (St. Louis, MO, USA). Ferulic acid (purity ≥98.0%) was obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). D. nobile was purchased from Omniherb (Yeongcheon, Korea). The origin of the sample was confirmed taxonomically by Professor Je-Hyun Lee, Dongguk University, Gyeongju, Republic of Korea. A voucher specimen (NO. KIOM-AO15) was deposited in storage at the Basic Herbal Medicine Research Group, Korea Institute of Oriental Medicine.
Preparations of DNE and standard solution
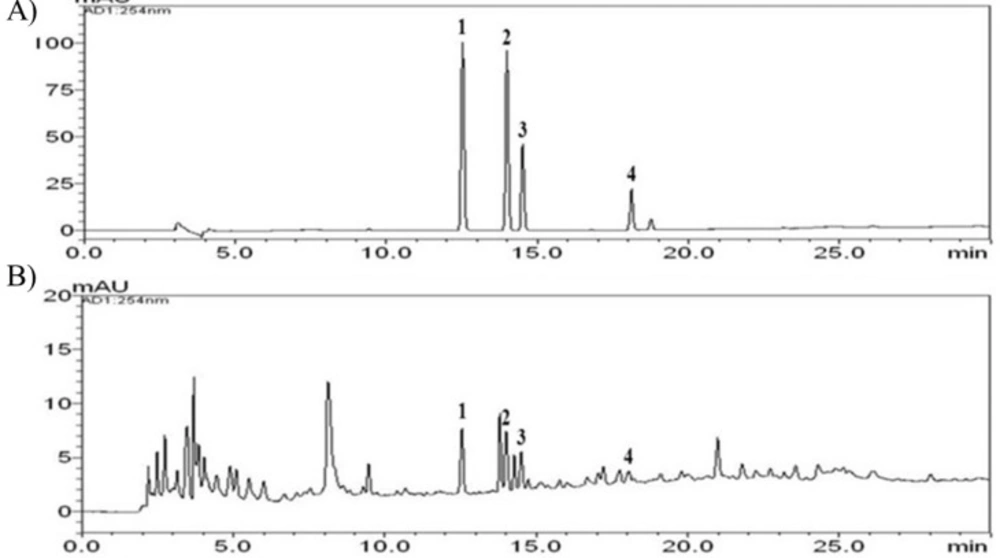
Dried D. nobile (60 g) was extracted with distilled water (600 mL) by reflux for 2 h. The extracted solution was filtered through filter paper, evaporated to dryness and freeze-dried (5.19 g). The yield of the water extract obtained was 17.5%. A lyophilized sample (20 mg) was dissolved in distilled water (10 mL) and mixed. The solution was filtered through a SmartPor GHP syringe filter (0.2 μm pore size, Woongki Science, Seoul, Korea). The stock solutions of the four reference standards were dissolved in methanol (1.0 mg/mL) and stored below 4°C.
HPLC analysis
A Shimadzu LC-20A HPLC system (Shimadzu Co., Kyoto, Japan) with a PDA detector was employed in this study. The data were processed using LCSolution software (Version 1.24; Shimadzu Co., Kyoto, Japan). The analytical column used for separation was a Gemini C18 column (250 × 4.6 mm; particle size 5 μm; Phenomenex, Torrance, CA, USA) and was maintained at 40°C. The mobile phases for chromatographic separation were carried out using a gradient elution of solvent A (1.0% v/v aqueous acetic acid) and solvent B (1.0% v/v acetic acid in acetonitrile). The gradient flow of the two-solvent system was as follows: 5% B (0 min), 5-70% B (40 min), 70-100% B (45 min), 100% B (50 min), 100-5% B (55 min), and 5% B (70 min). Analysis was performed at a flow-rate of 1.0 mL/min with a detection wavelength of 254 nm. The injection volume was 10 μL.
Cell viability assay
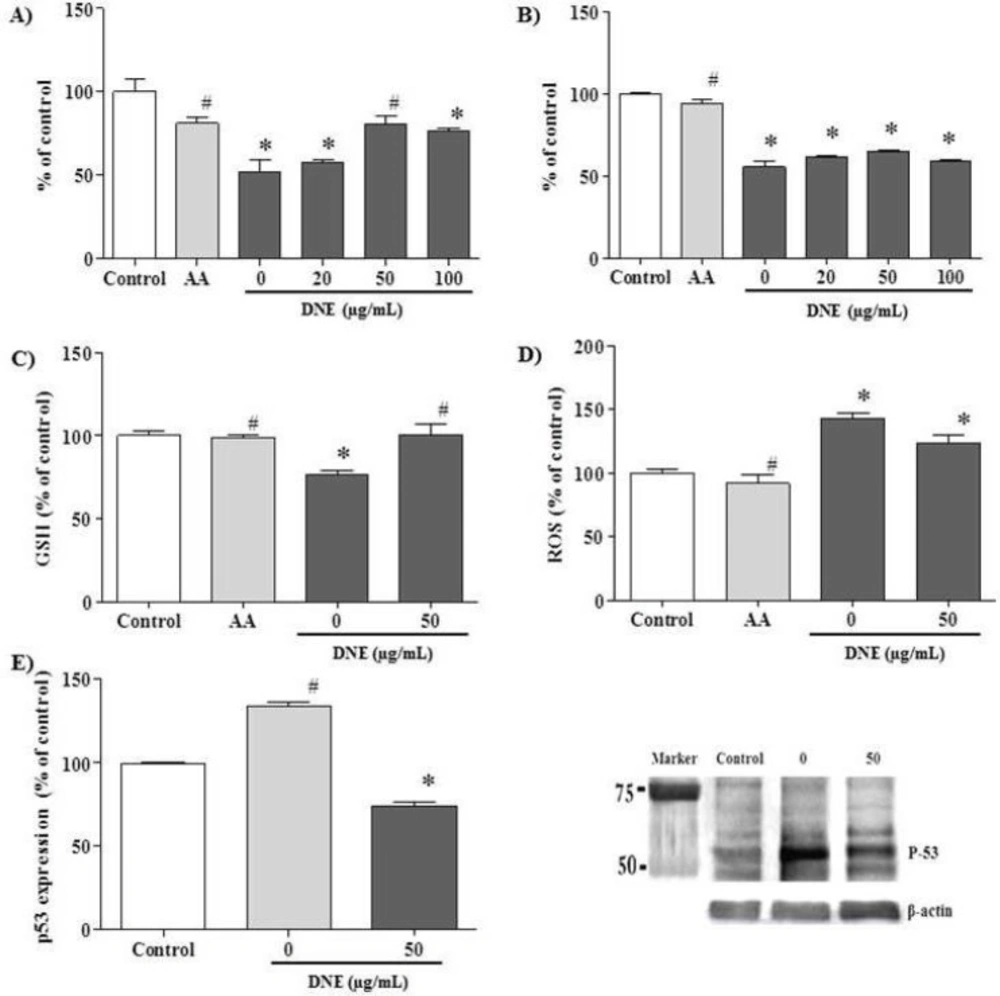
For all assays, PK15 cells were seeded at a density of 1 × 10
4 cells/mL in 96-well plates with regular growth medium. The experiments were carried out on the following day. The effect of DNE on the cisplatin-treated PK15 cells was assessed using MTT and crystal violet assays. PK15 cells were treated with ascorbic acid (positive control, 1.7 mg/mL) and DNE (0, 50, 100 and 200 µg/mL) 2 h before cisplatin (15 µg/mL) treatment; after cisplatin treatment, the cells were incubated for 24 h. The MTT assay was performed using the EZ-Cytox Cell Viability assay kit (Daeil, Seoul, Korea) according to the manufacturer’s protocol. The crystal violet assay was carried out based on a previous report with minor modifications (
17).
Reactive oxygen species and glutathione levels in PK15 cells
For both the reactive oxygen species (ROS) and the glutathione (GSH) assays, cells were pretreated with ascorbic acid (positive control, 1.7 mg/mL) and DNE (0 and 50 µg/mL). After 2 h, the cells were treated with cisplatin (15 µg/mL) and incubated for 24 h. The ROS contents were determined using the dihydrodichlorofluoroscein diacetate (Invitrogen, Carlsbad, CA, USA) method with some modification (
18). The level of GSH was determined with a Glutathione Assay Kit (Northwest, WA, USA) according to the manufacturer’s protocol.
Western blot of p53
PK15 cells were prepared in the same manner as described for the ROS and GSH assays. After incubation, the cells were lysed with lysis buffer (20 mM Tris-HCl pH 8, 150 mM NaCl, protease inhibitor cocktail). After the protein assay, a 30 µg protein sample was separated using 12% SDS-PAGE. The proteins were transferred onto a nitrocellulose membrane in a Semi-Dry Transfer system from Bio-Rad (Hercules, CA, USA). After blocking, the membrane was incubated with primary antibody anti-p53 (1:300; Santa Cruz Biotechnologies, Santa Cruz, CA, USA). Horseradish peroxidase-conjugated anti-mouse IgG (1:5000; Santa Cruz Biotechnologies) was used for p53 detection. Immunoreactivity was visualized using an enhanced chemiluminescence detection kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
Animal study
Four-week-old male Sprague Dawley (SD) rats were obtained from Orient Bio (Seongnam, Korea) and acclimated to laboratory conditions (25 ± 0.2°C, 50% relative humidity, 12 h light/dark cycle) for 1 week before experimentation. All animals were supplied with standard chow (Charles River Inc., Richmond, IN, USA) and water
ad libitum. Healthy SD rats (n = 30) were randomly allocated into 6 groups (n = 5/group) and treated for 28 days with each compound orally as follows: group 1 (control, distilled water; DW), group 2 (cisplatin alone, DW + cisplatin), group 3 (100 mg/kg of captopril + cisplatin), groups 4-6 (100, 300 and 500 mg/kg of DNE + cisplatin). Captopril, angiotensin-converting enzyme inhibitor, was used as a positive control drug, which is known to alleviate cisplatin-induced nephrotoxicity by its antioxidant properties and renin-angiotensin system inhibitory effect (
19,
20). On day 23, cisplatin (5 mg/kg) was injected intraperitoneally to induce AKI, except for the control group. On day 27, the urine of each animal was collected over 24 h for urine volume analysis; 3 h after the last treatment, the animals were anesthetized with an intraperitoneal injection of a combination of zolazepam and tiletamine (Zoletil; Virbac). The experimental protocols were approved (No. CNU-00070) by the Institutional Animal Care and Use Committee of Chungnam National University (Daejeon, Korea). Blood samples were collected from the inferior vena cava and separated by centrifugation at 800
g for 15 min, and the serum blood urea nitrogen (BUN) and creatinine (CRE) were determined on a dry chemistry system (IDEXX Laboratories, Westbrook, ME, USA). The left kidney was quickly removed for histopathological analysis. The other kidney was removed and used in the GSH (Northwest, WA, USA) and the malondialdehyde (MDA; Northwest, WA, USA) assays. The assays were conducted according to the manufacturer’s protocol.
Histopathological examination
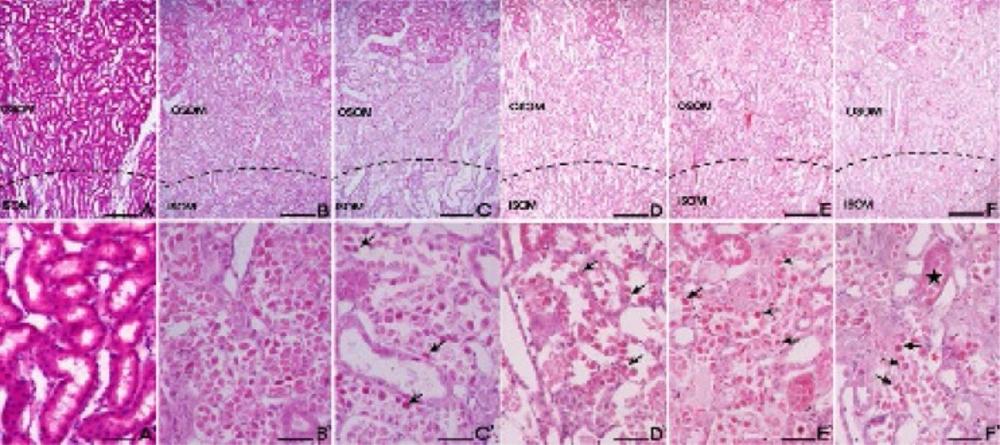
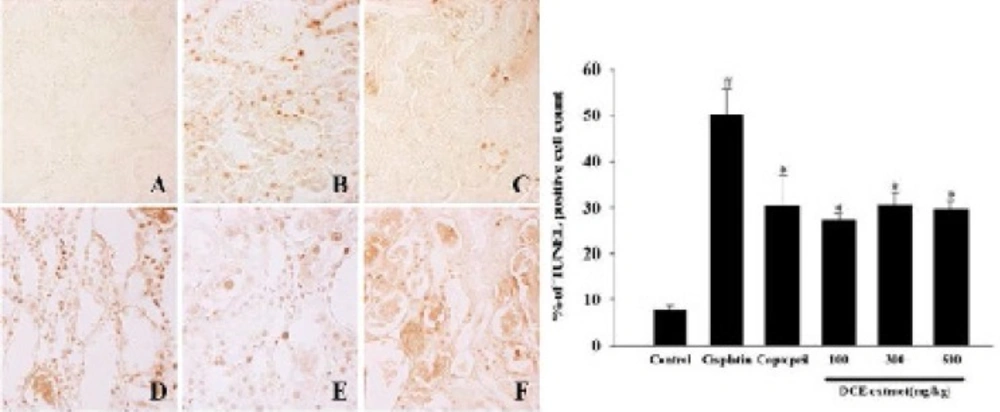
The left kidney was fixed immediately in a 10% buffered formalin phosphate solution, embedded in paraffin and cut into 5 μm sections and processed for histological staining. These serial tissue sections were either stained with haematoxylin and eosin (H&E) for histopathological examination or subjected to TUNEL staining. Apoptotic nuclei were detected by the TUNEL method using an apoptosis detection kit (Millipore, Bilerica, MA, USA) according to the manufacturer’s protocol. All of the stained slides were analyzed under the light microscope.
Statistical analysis
Data are presented as mean values ± standard error of mean (SEM). Significant differences among the experimental groups were determined using the one-way analysis of variance (ANOVA) test followed by Tukey’s post hoc analysis. Values of P < 0.05 were considered to be statistically significant.