Chemicals and Cell lines
All the materials and reagents for archaeal cultures were purchased from Merck (E. Merck, Darmstadt, Germany). Ethyl acetate for SM extraction was purchased from Samchun (Korea). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), Dulbecco’s modified Eagle’s medium (DMEM) with high glucose, FBS (Fetal Bovine Serum), trypsin, penicillin, and streptomycin were purchased from Biosera (Austria). Ultra-purified water was used throughout the analysis and all other chemicals were analytical grade.
Prostate cancer cell lines (PC3 and DU145) were obtained from national cell bank of Iran (Pasteur Institute of Iran, Tehran, Iran). Breast cancer cell lines of MDA-MB-468 (IBRC C10095) and MCF-7 (IBRC C10082) and lung cancer cell line (A549; IBRC C10080), were obtained from cell bank of Iranian Biological Resource Center, Tehran, Iran. Human foreskin fibroblast (HFF-5) cell line was obtained from Royan Institute stem cell bank.
Archaea culture and supernatant isolation
Eight strains from halophilic archaeal strains were selected. The archaeal strains were obtained from IBRC microorganisms bank (
Table 1). All strains were cultured in Modified Growth Medium (MGM) with 23% (w/v) of total salt (
14). For SM production, a loopful of agar slant culture of each halophilic archaeal strain was inoculated into 25 mL of MGM. The inoculated medium was incubated at 40 °C for 48 h on a rotary shaker at 150 rpm, and then transferred into 225 mL medium to cultivate at the same temperature for 7 days.
Metabolite extraction from supernatant
The archaeal culture medium was removed and centrifuged at 4000 g for 40 min. The cell-free supernatant was collected after centrifugation and was filtered with Whatman no.1 filter paper. The equal volume of ethyl acetate was added to the supernatant with the ratio of 1:1 and it was shaken well for 2 h at room temperature. Then, it was allowed to settle overnight in a stand, at 4 °C. The upper organic layer containing SM was collected into a clean and dry bottle and was evaporated by BUCHI Rotavapor R_114 (Switzerland) until 1 mL of total volume remained in the rotary balloon. The remaining volume was transferred to a sterile vial with an identified weight and later on, it was dried completely by rotary. The dried SM was weighted and the pure weight of the SM was calculated. Approximately 3.5 mg of SM were obtained from 1 L of each archaeal culture. The SM was dissolved in DMSO (Merck, Germany), serving as a stock solution, which was later diluted to a final solvent concentration. The SM was tested as the weight of total SM/volume (
11).
Human Cell culture
Human prostate cancer cells (DU145, PC3), breast cancer cells (MDA-MB-468, MCF-7), lung carcinoma (A549) and human foreskin fibroblast cells (HFF-5 as normal control) were grown as a monolayer in DMEM supplemented with 10% (v/v) fetal bovine serum (FBS), 1.0 mM sodium pyruvate and 2 mM L-glutamine,100 U/mL penicillin and 100 μg/mL streptomycin at 37 °C in a humidified atmosphere of 95% air and 5% CO2.
MTT assay
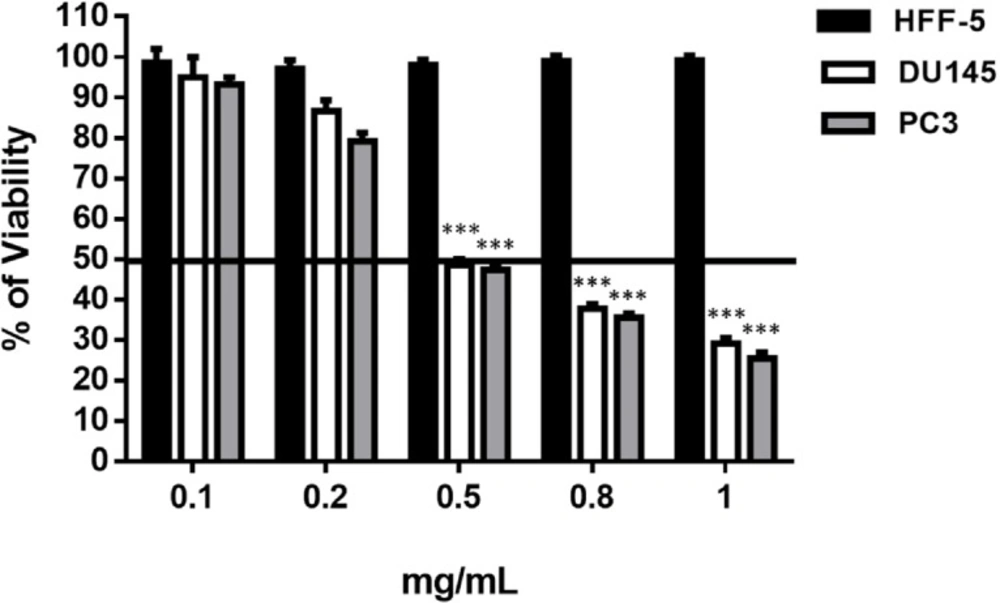
The MTT-assay was used to evaluate the effect of the SM on cell viability pre and post treatment. Briefly, 5000 cells/well were seeded onto a 96-well plate and were allowed to adhere for overnight. Cultivated cells were incubated with different concentrations of SM (0, 0.01, 0.1, 0.2, 0.4 and 0.8 mg/mL) for 48 h. The final concentration of DMSO was <1 mM that was not cytotoxic for the cells. To assess the viability of the cells, 10 μL of 5 mg/mL solution of MTT in PBS was added to each well and incubated for 3 h at 37 °C. Finally, the medium was removed and 100 μL of the DMSO was added to each well to solubilize the blue formazan. Dye absorbance was measured at 560 nm (
13). One chamber of each 96-well plate contained only DMSO and its OD
560 assumed as blank. All of the ODs (control and treated groups) from each plate first subtract from the blank of the same plate and then the mean of OD
560 for control and treated groups were calculated. The percentage of viable cell calculate via mean OD
560 (Treated group)/mean OD
560 (Control group) × 100.
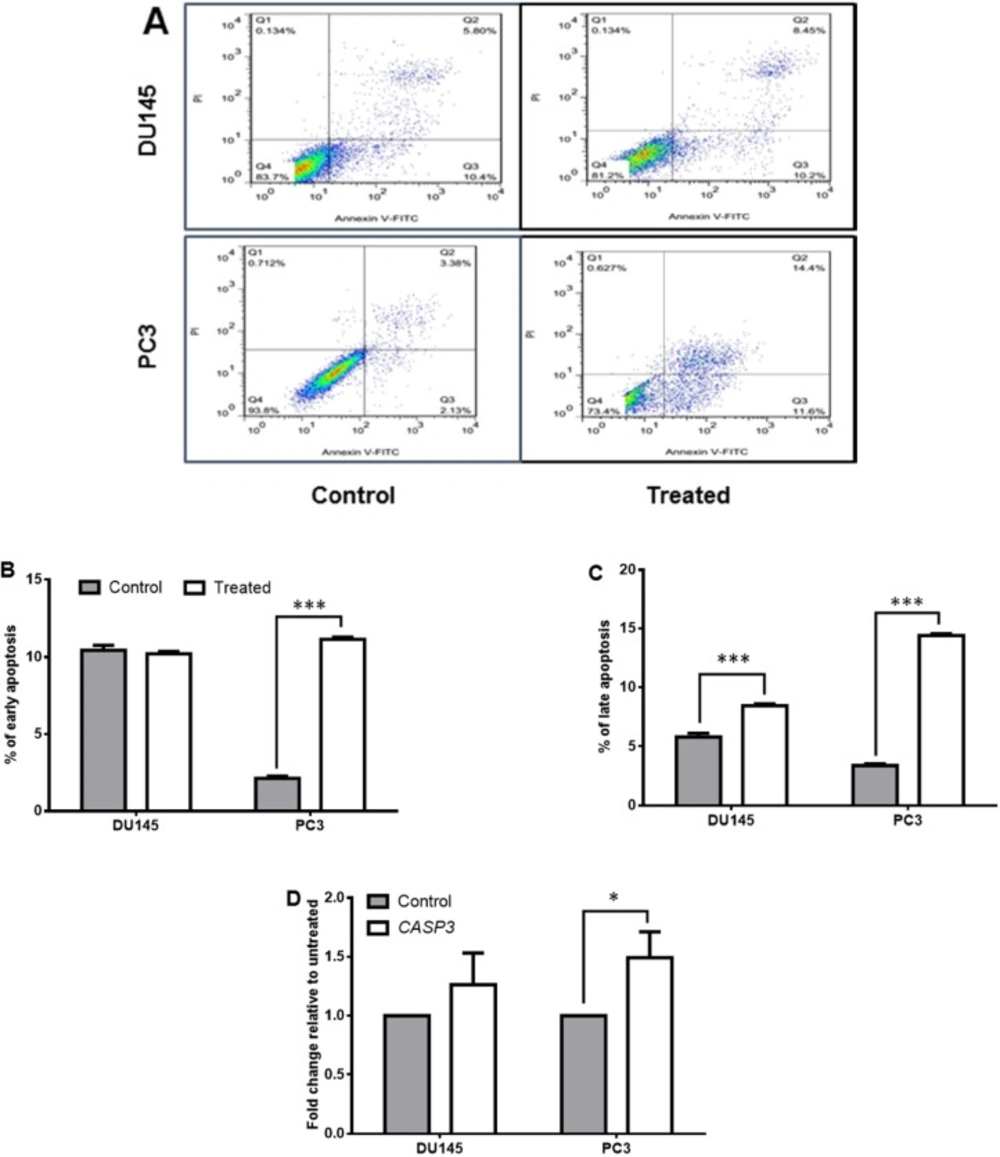
Apoptosis test
The apoptotic effects of SM on treated and control cells were measured using the AnnexinV/PI Staining Kit (Sigma) according to the manufacturer’s instruction. Binding of annexin-V to phosphatidylserine suggests early apoptosis, whereas binding to both annexin-V and PI indicates late apoptosis.
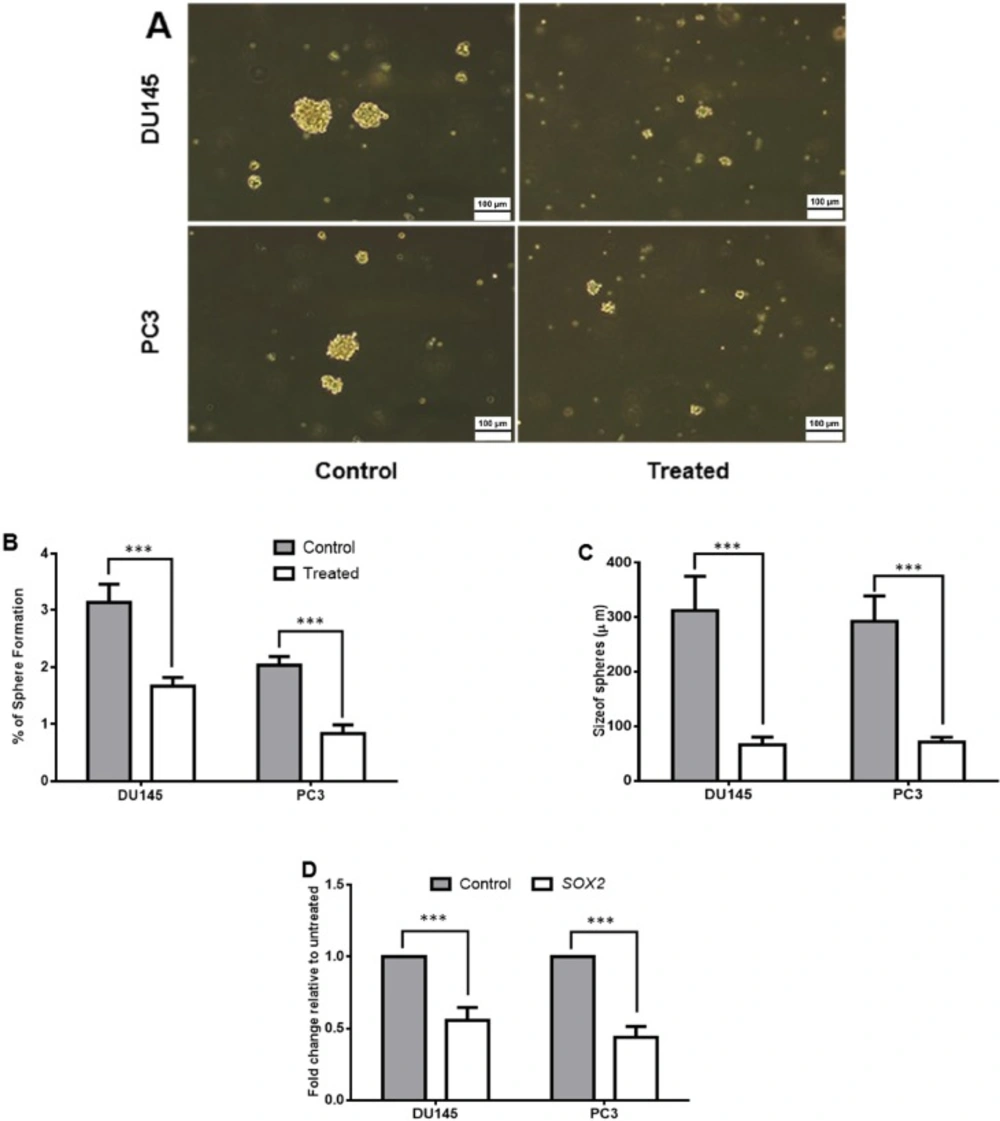
Sphere formation assay
Sphere formation capacities were assessed after 48 h of metabolite pretreatment. To do this first, the cells were seeded at a density of 106 cells/well in 6-well plate and allowed to adhere to plate for 24 h. Then they were treated by the SM IC50 dose for 48 h. Un-treated cells were used as control group. Then the cells were harvested and followed to sphere formation assay. Briefly, 500 cells/cm2 were seeded in culture dishes coated with (2-hydroxyethyl methacrylate) (poly-HEMA, Sigma) and cultivated in serum free DMEM supplemented with B27 (GIBCO, Karlsruhe, Germany), 20 ng/mL EGF and bFGF (Royan Institute, Tehran, Iran) and PenStrep. The cells were grown for 7 days and maintained in a humidified incubator at 37 °C and an atmospheric pressure of 5% (v/v) carbon dioxide/air. Then, the spheres with diameters of >50 µm were counted using an eyepiece graticule. The percentage of plated cells which formed sphere was calculated, and was referred as the percentage of sphere formation. The values were expressed as means ± SD of at least three independent experiments.
Quantitative real-time RT-PCR (qRT-PCR)
Total mRNA was isolated from treated and control cell lines after 48 h using TRIzol Reagent (Invitrogen) according to the manufacturer’s instruction. cDNA was synthesized using the RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas, Waltham, Massachusetts, USA). SYBR Premix Ex Taq II (Tli RNase H Plus) (Takara, Berkeley, California, USA) was utilized to perform quantitative real-time reverse transcription-PCR (RT-PCR) using Rotor-Gene 6000 (Corbett). The
GAPDH gene transcript was measured as a normalizer to determine the other gene relative transcripts (2
-ΔΔCt). The sequences of primers are listed in
Table 2.
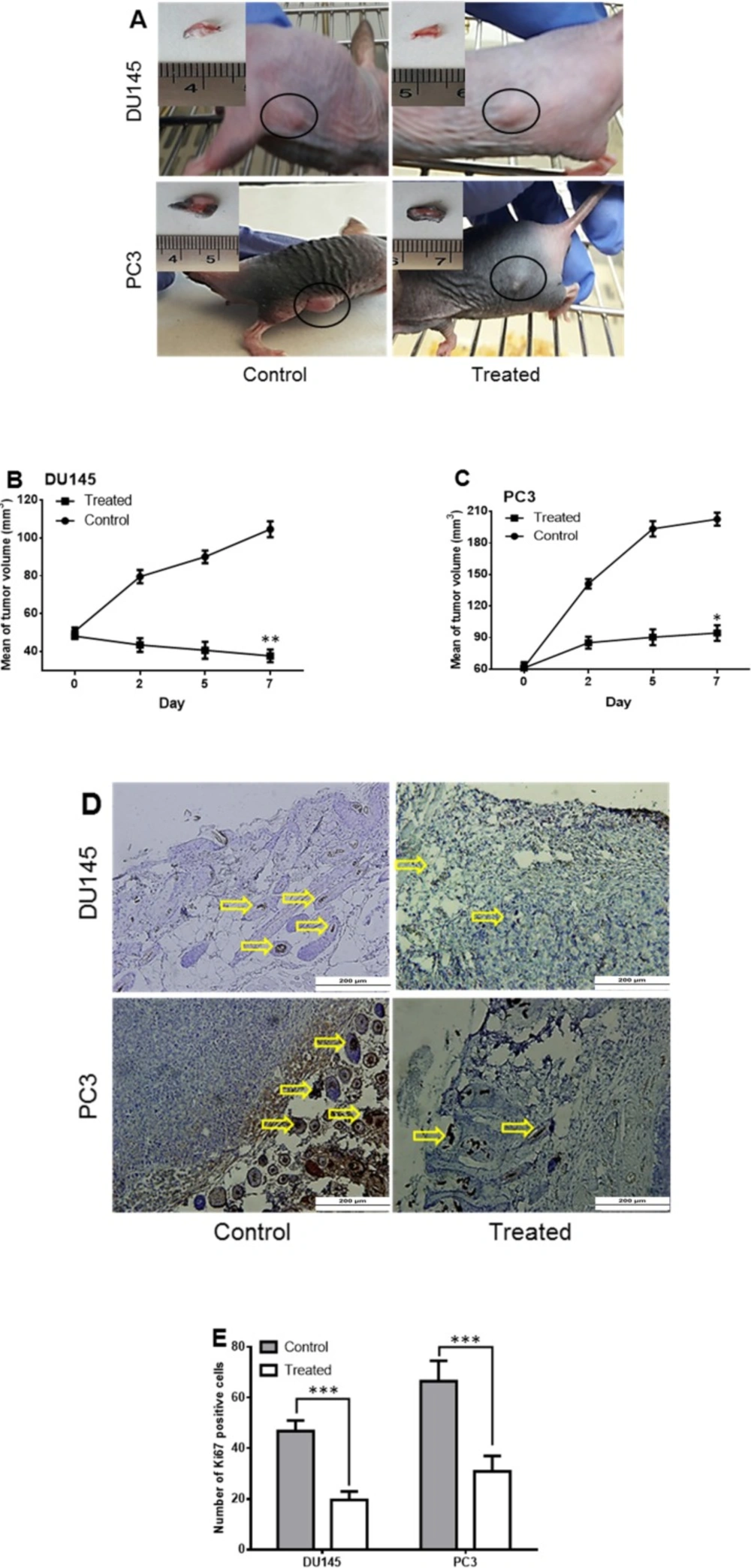
Mouse Tumorigenicity Assay
The suspension of 3 × 10
6 cells (PC3, DU145) was prepared in 20 µL of matrigel (0.34 mg/mL, BD Bioscinces, San Jose, CA, USA). Six Nude female mice in the ages of 4-6 weeks (were purchased from Pasteur Institute of Iran and maintained under clean area provided by HEPA-Filtered Laminar-Air-Flow Systems). The animals were allowed to acclimate for two weeks prior to experimentation. All mice were injected with the 3 × 10
6 cells subcutaneously, one injection on the flank in each animal (six mice with PC3 and six with DU145). Once tumors got palpable (about 3 days post injection), tumor volumes were calculated using the formula: (
L ×
W2)/2 which L and W indicated length and width, respectively. The length and width were measured with a caliper every other day. To find the proper dose of SM intra-tumor injection we considered the 0.5 mg/mL dose of
in-vitro injection as 500 mg/kg (
15,
16). Because the minimum weight of our mice was 17.5 g, we injected 8.75 mg of SM intratumorally in each mouse in the treated group. We select the minimum dose to avoid the death of mice. The SM was dissolved in DMSO (total volume 70 µL) and then intra-tumor injection was done. The tumors of one PC3 and one DU145 injected mice were injected with only DMSO (70 µL) as the control. The mice were sacrificed in a humane manner when the tumor size exceeds 25 mm in diameter in either direction.
Immunohistochemistry
Isolated tumors were fixed by paraformaldehyde (4% V:V) and then horizontal serial sections at 4 mm thickness were prepared by using a Leica RM2125RT microtome (Leica RM2125RT, Leica Microsystems Inc., Bannockburn, IL). For immunohistochemically assays, the sections were deparaffinized, and subjected to antigen retrieval (antigen retrieval PH9, Dako) for 30 min. Sections were rinsed thoroughly with phosphate-buffered saline containing 0.05% tween-20 (PBST) and endogenous peroxidase activity was quenched with 1% H2O2 in methanol for 30 min in the dark. To increase the permeability, the sections were treated with Triton X-100 (0.5%) for 10 min and then blocked with 10% goat serum for one hour (
17). Then, the slides were incubated with primary antibody human Ki67 antibody (1:100, Santa Cruz Biotechnology, INC., USA) at 4 °C, overnight; afterwards, the slides were washed with PBST and incubated for one hour at 37 °C with horseradish peroxidase (HRP) conjugated secondary antibodies (Abcam6789 1:500 dilution). Finally, color was developed by treating samples with 3, 3›-diaminobenzidine (DAB; Sc-2051, Santa Cruz) for 5 min. The sections were then washed with distilled water, counterstained with hematoxylin, dehydrated with two changes of alcohol, cleared by xylene and cover slipped. The labeled cells were visualized using Olympus BX51 microscope and images were captured with an Olympus DP70 digital camera under the same setting. Measurement of positive Ki67 positive cells was performed in ten randomly selected areas of five pictures from three different replicates with an eyepiece graticule. The means of the different experimental groups were compared by ANOVA. The differences were considered to be statistically significant at
p < 0.05.
Statistical analysis
One-way ANOVA test was used to determine the statistical significance of the differences between the treated and the control groups. All of the experiments were performed in triplicates or more and the data obtained were expressed as means ± SD. p < 0.05 was considered as statistically significant.