3.1. Medicinal Materials and Preparation
The formulations of BTD and BHD are presented in Table S1 in the Supplementary File. All raw herbal materials were obtained from the Traditional Chinese Medicine Pharmacy at Xi'an Hospital of Traditional Chinese Medicine and were identified as meeting the legal standards for the corresponding varieties in the Pharmacopoeia of the People's Republic of China (2020 edition, Volume I). Each batch of herbal decoction was standardized by yield and concentration.
For preparation, BTD and BHD were soaked in 500 mL of purified water (grade III) for 30 minutes. Additional purified water (grade III) was added at a weight-to-volume ratio of 1:8. The medicinal herbs and water were placed into a decoction machine and boiled 3 times for 1.5 hours, 1 hour, and 45 minutes, respectively. After the decoction solutions were combined, they were concentrated and precipitated overnight with ethanol. After filtration and ethanol distillation, the preparations were concentrated and stored in 500-mL wide-mouth reagent bottles at 4°C in a refrigerator.
Methodological validation of astragaloside IV content determination in the BTD compound preparation was completed. The method met the requirements for specificity, linearity, repeatability, accuracy, intermediate precision, and robustness. The average astragaloside IV content in BTD was 0.68 mg/g. Batch consistency was controlled based on astragaloside IV content.
The dosage was calculated using the body surface area method for equivalent dose conversion between humans and rats. A standard adult body weight of 70 kg and a rat body weight of 200 - 250 g were used as baselines. A human-to-rat dose conversion factor of 6.25 was applied in combination with the clinical adult dosage. The dosage volume for the BTD group was 0.9 mL/250 g per administration, and that for the BHD group was 0.675 mL/250 g per administration.
3.2. Preparation of the Acute Ischemic Stroke Model
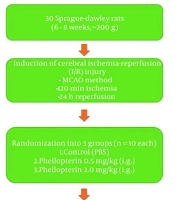
A rat model of focal cerebral ischemia was induced using an adapted filament occlusion technique targeting the right hemisphere. Animals were anesthetized by intraperitoneal injection of ketamine (70 mg/kg) combined with xylazine (5 mg/kg) and then placed in the dorsal recumbent position on a thermoregulated heating platform to maintain core body temperature at 37°C throughout the operation. Postoperative analgesia was provided by administering buprenorphine (0.05 mg/kg, subcutaneously) every 12 hours for the first 48 hours after surgery.
After antiseptic preparation of the cervical region, a 2-cm midline skin incision was made. Through blunt dissection, the submandibular gland fascia was separated to expose the right sternocleidomastoid and sternohyoid muscles. The intermuscular space was carefully dissected to visualize the carotid sheath. Under microscopic guidance, the right common carotid artery (CCA) was meticulously isolated while preserving the adjacent vagus nerve. Subsequent dissection exposed the internal carotid artery (ICA) and external carotid artery (ECA). Temporary vascular occlusion was achieved by clamping the ICA, while simultaneous occlusion was applied to both the ECA and the proximal segment of the CCA.
The CCA was carefully ligated, and a small V-shaped incision was made in its wall. A suture thread was then introduced into the ICA through the CCA and carefully advanced toward the skull until it reached the origin of the middle cerebral artery. The optimal insertion distance was approximately 18 - 20 mm from the bifurcation of the internal and external carotid arteries, and advancement was stopped when mild resistance was encountered. The suture plug was ligated and fixed, the excess suture plug was removed, streptomycin was sprinkled on the surgical area, and the muscle and skin were sutured layer by layer.
Throughout the procedure, a laser speckle imaging system continuously monitored blood flow changes in the affected brain region, and a substantial reduction in cerebral perfusion was used as the key indicator of successful model establishment. At 1.5 hours after surgery, the suture plug was removed for reperfusion, and subsequent experiments were conducted 24 hours after reperfusion.
The following exclusion criteria were applied: 1) subarachnoid hemorrhage confirmed by postmortem inspection; 2) Longa score < 1 at 24 hours after modeling; and 3) death during surgery. All animals were assessed for these criteria immediately after surgery and at 24 hours. In the model group, 2 animals met criterion 1 and were excluded before any further analysis; they were replaced by new animals that underwent the same modeling procedure to maintain n = 12 for neurological scoring.
Humane endpoints were predefined as follows: 1) body weight loss exceeding 20% of baseline; 2) inability to access food or water; and 3) a moribund state characterized by lethargy, hypothermia, and labored breathing. Animals reaching any of these endpoints were immediately euthanized by pentobarbital sodium overdose (200 mg/kg, intraperitoneally).
3.3. Experimental Animals and Grouping
Male Sprague-Dawley rats (6 - 8 weeks old; body weight, 200 - 250 g) certified as specific pathogen-free were obtained from Shaanxi Pharmaceutical Medical Biotechnology Company. The animals were housed under controlled environmental conditions (22 ± 2°C, 12-hour light/dark cycle), with free access to food and water during a 7-day adaptation period.
Allocation concealment was achieved using sequentially numbered, opaque, sealed envelopes prepared by an independent researcher who was not involved in animal surgery or outcome assessment. The randomization sequence was generated using SPSS version 26.0 (IBM Corporation, USA) and was kept concealed until group assignment. Animal welfare monitoring was performed twice daily (8:00 AM and 8:00 PM) throughout the experimental period. Body weight, food and water intake, general appearance, and neurological status were observed.
The experimental design included the following treatment conditions (n = 12):
1) Sham group: Rats underwent skin incision and suture insertion without vessel ligation, mirroring all other operative steps performed in the model group. Oral administration of normal saline was initiated on the second day after surgery, twice daily for 2 consecutive weeks.
2) Model group: A focal cerebral ischemia-reperfusion model was prepared using an improved intraluminal suture method. Starting on the second day after surgery, normal saline was administered orally twice daily for 2 consecutive weeks.
3) BHD treatment group: Oral administration of BHD was initiated on the second day after surgery, 3 times daily for 2 consecutive weeks at a dosage volume of 0.675 mL/250 g/time.
4) BTD treatment group: Oral administration of BTD was initiated on the second day after surgery, twice daily for 2 consecutive weeks at a dosage volume of 0.9 mL/250 g/time.
The entire outcome evaluation process was conducted in a blinded manner. For endpoint-specific assays, including cerebral blood flow, infarct volume, Western blotting, and enzyme-linked immunosorbent assay (ELISA), animals were randomly subsampled from each group using a separate computer-generated randomization list to ensure that the animals selected for different assays were representative of the whole cohort. Staff responsible for neurological function scoring, cerebral blood flow measurement, infarct volume analysis, histological evaluation, transmission electron microscopy observation, Western blot quantification, and ELISA detection were unaware of the group assignments.
For neurological function scoring, 12 animals from each group were tested. For cerebral blood flow measurement, 5 animals were randomly selected from each group of 12. For infarct volume, Western blotting, and ELISA, 3 animals were randomly selected from each group of 12.
The research procedures were approved by the Experimental Animal Ethics Committee of Shaanxi University of Chinese Medicine (Approval No. SNCMDL20250903003), and all animal experiments were conducted in full compliance with China's established standards for laboratory animal welfare and use. This investigation adhered to the updated ARRIVE 2.0 experimental guidelines (
21).
3.4. Evaluation of Neurological Dysfunction Using the Longa Score
The neurological status of all experimental rodents was assessed using the Longa scale at 2 time points: 24 hours after model establishment and 14 days after treatment initiation. Higher scores indicated greater neurological dysfunction. The scoring system was as follows: 0, normal neurological function; 1, observable flexion and adduction of the contralateral forelimb when the tail was elevated; 2, circular movement toward the affected side during locomotion; 3, loss of balance toward the impaired side while moving or standing; and 4, complete absence of voluntary movement accompanied by impaired consciousness.
Two animals from the model cohort were eliminated from the study because of subarachnoid hemorrhage. These animals were replaced with additional rats to maintain n = 12 per group for neurological scoring and mortality-sensitive outcomes.
3.5. Detection of Cerebral Cortical Blood Flow
The rats were anesthetized using ketamine (70 mg/kg) and xylazine (5 mg/kg) before being secured on the surgical platform. A cutaneous incision was made over the cranial region of interest to expose the underlying bone structure. The laser-based microcirculation monitoring device (Moor Instruments, UK) was carefully positioned on the exposed cranial surface, with deliberate avoidance of the midline suture, to measure regional cerebral cortical perfusion.
3.6. Quantification of Cerebral Infarct Volume by TTC Staining
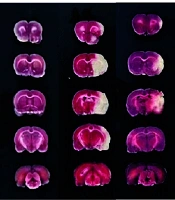
At the end of the experimental procedures, the rats underwent transcardiac perfusion with chilled phosphate-buffered saline (PBS). The animals were then euthanized by decapitation, and their brains were promptly excised and placed in a -20°C environment for 30 minutes. The cryopreserved cerebral tissue was subsequently removed and precisely divided into 5 equidistant coronal sections, each 2 mm thick, while maintained on ice. All sections were immersed in 2% 2,3,5-triphenyltetrazolium chloride (TTC) solution (Sigma-Aldrich, USA) and incubated in the dark at room temperature for 20 minutes. After staining, the tissue samples were preserved in 4% paraformaldehyde and systematically arranged for photographic documentation. Viable neural tissue appeared red, whereas ischemic regions remained unstained and appeared pale white. Cerebral infarction was quantified using digital image analysis with ImageJ software (NIH, USA), and the infarct percentage was determined using the following equation: Percentage of cerebral infarction = (Aggregate volume of infarct region across all sections / Total sectional brain volume) × 100%.
3.7. Nissl Staining
At the end of the experimental procedures, the rodents underwent transcardiac perfusion with chilled PBS. The brain was then extracted, with a specific focus on obtaining ischemic hippocampal regions. Tissue blocks were fixed in 4% paraformaldehyde phosphate buffer for 15 minutes, followed by preparation of 10-μm paraffin sections through dehydration, clearing, and wax immersion. The prepared sections were first washed under running water and then stained with Nissl staining reagent (Solarbio, China) for 15 minutes. After staining, the sections were gently rinsed with distilled water. Rapid dehydration was then performed using ascending concentrations of absolute ethanol, followed by xylene clearing for 5 minutes. The sections were sealed with neutral gum and observed under an optical microscope (Olympus, Tokyo, Japan) for image acquisition and analysis.
3.8. Hematoxylin and Eosin Staining
Paraffin-embedded tissue samples were first dewaxed using Eco-Friendly Dewaxing Agents I and II, with 20 minutes of incubation in each solution to ensure complete paraffin removal. The samples were then progressively rehydrated through an ethanol gradient consisting of absolute ethanol I and absolute ethanol II, each for 5 minutes, and 75% ethanol for 5 minutes, followed by extensive washing under running tap water.
After hydration, the sections were treated with high-definition staining pretreatment solution for 60 seconds and then immersed in hematoxylin dye for 3 - 5 minutes. After rinsing with tap water, the sections were differentiated with differentiation solution, rinsed again with tap water, blued with blue solution, and rinsed under running water. The sections were then briefly dehydrated in 95% ethanol for 1 minute and counterstained with eosin for 15 seconds. Final dehydration was performed using absolute ethanol twice for 2 minutes each, and the sections were cleared in n-butanol twice for 2 minutes each to enhance transparency. The tissue samples then underwent sequential immersion in ethanol solutions of increasing concentrations (70%, 80%, 90%, and 100%, each for 15 minutes), followed by 2 xylene treatments for 2 minutes each. Finally, the sections were sealed with neutral gum and examined under an optical microscope (Olympus, Tokyo, Japan) for image acquisition and analysis.
3.9. Transmission Electron Microscopy
Fresh infarcted brain tissue was immediately dissected into 1-mm3 pieces after harvest and fixed in precooled 2.5% glutaraldehyde fixative at 4°C for 4 hours. After 3 rinses with 0.1 M PBS, the tissue pieces were fixed in 1% osmium tetroxide solution (Sigma-Aldrich, USA) in the dark at room temperature for 2 hours. Subsequent processing involved sequential dehydration through increasing ethanol concentrations (50%, 70%, 80%, 90%, and absolute ethanol, each for 15 minutes), followed by propylene oxide treatment, progressive epoxy resin infiltration, and final embedding polymerization. Ultrathin sections measuring 70 nm were prepared using an ultramicrotome. These sections underwent double staining with uranyl acetate in ethanol and lead citrate solution before examination and documentation using a transmission electron microscope (Hitachi High-Tech, Japan).
3.10. Western Blotting
Brain specimens were carefully weighed and mechanically disrupted in ice-cold RIPA buffer containing 1% protease inhibitor and 2% phosphatase inhibitor at a 1:10 tissue-to-buffer ratio until complete dissolution was achieved. After homogenization, the mixture was centrifuged at 4°C and 12,000 rpm for 10 minutes, and the resulting supernatant was collected as the protein extract. Protein quantification was performed using a BCA assay kit (Beyotime, China), after which samples were heat-denatured at 100°C for 10 minutes for subsequent analysis.
The prepared protein samples were separated by SDS-PAGE and then transferred onto PVDF membranes (Merck Millipore, USA). Membranes were blocked with 5% nonfat milk at ambient temperature for 60 minutes. Primary antibodies against NLRP3 (BosterBio, China), ASC (Bioss, China), pro-Caspase-1 (Abcam, USA), cleaved Caspase-1 (Affinity Bioscience, USA), GSDMD (Biodragon, China), N-GSDMD (HuaBio, China), and HIF-1α (Bioss, China) were added, and the membranes were incubated overnight at 4°C. Secondary antibodies were incubated with the samples at ambient temperature for 60 minutes. Chemiluminescent detection was then performed using immunoblotting reagents to visualize protein bands. The resulting bands were captured digitally and subjected to quantitative analysis using ImageJ software (NIH, USA), with band intensity measurements providing semiquantitative data.
3.11. Enzyme-Linked Immunosorbent Assay
The concentrations of inflammatory mediators, including IL-1β, tumor necrosis factor-alpha (TNF-α), IL-18, and HIF-1α, were quantified using commercial ELISA kits (Beyotime, China) according to the manufacturer's protocols. The assay procedure involved processing brain tissue homogenate supernatants and serum samples through sequential steps, including sample application, incubation, plate washing, reagent addition, chromogenic reaction, and termination. Optical density measurements were recorded at 450 nm.
3.12. Statistical Analysis
Statistical analyses were conducted using GraphPad Prism software (version 10.1.2), and results are expressed as mean ± standard deviation (mean ± SD). Before one-way analysis of variance (ANOVA) was used, the normal distribution of the data was verified using the Shapiro-Wilk test, and variance equality was examined using Levene's test. The dataset satisfied both normality and variance homogeneity prerequisites. For repeated measures, including neurological scores at 24 hours and 14 days, 2-way repeated-measures ANOVA was applied, with group and time as factors. For intergroup mean comparisons, ANOVA was used, supplemented by Dunnett post hoc analysis for specific group contrasts. Statistical significance was defined as P < 0.05.
The primary outcomes were neurological function, assessed using the Longa score, and cerebral infarct volume, assessed using TTC staining. Secondary outcomes included cerebral blood flow, histological changes using Nissl and H&E staining, ultrastructural findings using transmission electron microscopy, pyroptosis-related protein expression using Western blotting, and inflammatory cytokine levels using ELISA.







 Neurological scores assessed 24 hours after model establishment, before administration. (B) Neurological scores assessed 2 weeks after administration. Data are presented as mean ± SD (n = 12). Statistical significance was defined as P < 0.05. BTD, Buyang Tongluo Decoction; BHD, Buyang Huanwu Decoction.")

 Representative TTC-stained coronal brain sections show the infarct area (white) in each group. (B) Quantitative analysis of cerebral infarct volume. Data are presented as mean ± SD (n = 3). Statistical significance was defined as P < 0.05. BTD, Buyang Tongluo Decoction; BHD, Buyang Huanwu Decoction.")
. BTD, Buyang Tongluo Decoction; BHD, Buyang Huanwu Decoction.")
. BTD, Buyang Tongluo Decoction; BHD, Buyang Huanwu Decoction.")
 Representative Western blot bands show the expression levels of NLRP3, ASC, pro-Caspase-1, cleaved Caspase-1, GSDMD, N-GSDMD, and HIF-1α. (B) Quantitative analysis of protein expression levels normalized to GAPDH. Data are presented as mean ± SD (n = 3). Statistical significance was defined as P < 0.05. BTD, Buyang Tongluo Decoction; BHD, Buyang Huanwu Decoction.")
. BTD, Buyang Tongluo Decoction; BHD, Buyang Huanwu Decoction.")
 Levels of IL-1β, TNF-α, IL-18, and HIF-1α in serum and (B) brain tissue homogenates measured by ELISA. Data are presented as mean ± SD (n = 3). Statistical significance was defined as P < 0.05. BTD, Buyang Tongluo Decoction; BHD, Buyang Huanwu Decoction.")